host genetic factors in colorectal cancer...
TRANSCRIPT

HOST GENETIC FACTORS IN COLORECTAL CANCER
METASTASIS
Anna Kinio
Department of Microbiology and Immunology, McGill University, Montreal
August 2014
A thesis submitted to McGill University in partial fulfillment of the requirements of the degree of M.Sc.
Microbiology and Immunology
© Anna Kinio, 2014

2
ABSTRACT
Colon cancer is the fourth most prevalent cause of death in female and male cancer patients and
is the third most frequent disease in the developed world. The metastasis of cells from the
primary tumor is directly related to poor patient prognosis and accounts for 90% of colon cancer
deaths. In the past, research efforts have focused on defining the genetic and molecular
mechanisms of tumor cells, however, it is now apparent that host processes are important in
determining cancer growth and metastatic outcome. Factors such as the interaction between the
tumor and its microenvironment within the host, tumor immune surveillance and configuration
of the vasculature play a primary role in determining tumor growth, homing to distant sites and
organ specificity of metastasis. Immunoediting of cancer contributes to host resistance and
provides selective pressure which ultimately determines the outcome of the disease. Further, the
metastatic niche secretes effectors to ready it for cancer cell colonization and metastatic growth
and dissemination. Our laboratory has implemented the use of both forward and reverse genetic
platforms to address the vast challenge of characterizing the host genetic basis of metastasis
resistance. While our candidate gene approach, investigating specific genes with key functions in
cell death and/or innate immunity did not yield significant results, our phenotype-driven
screening of chemically mutagenized mice, using N-ethyl-N-nitrosourea [ENU] germline
mutagenesis, led to the identification of two mouse pedigrees that showed resistance to colorectal
cancer metastasis to the lung, as assessed by mouse survival. Whole genome exome sequencing
revealed potential mutations in Nbeal1, Cadm3, and Ap1g2, which may be conferring the deviant
phenotype. Identifying the mutation underlying metastasis resistance in these pedigrees will lead,
not only to a more comprehensive understanding of the pathogenesis of cancer progression, but
may also provide opportunities for the development of novel therapeutic avenues for the
treatment of cancer metastasis.

3
RÉSUMÉ
Le cancer du colon est la troisième maladie la plus fréquente dans le monde développé et la
quatrième cause la plus fréquente de décès chez les patients atteints de cancer. La métastase des
cellules cancéreuses à partir de la tumeur primaire est directement liée à un mauvais pronostic et
est responsable de 90% des décès suite à un cancer du côlon. Dans le passé, les efforts de la
recherche se sont concentrés à définir les mécanismes génétiques et moléculaires ayant lieu dans
les cellules cancéreuses pour leur permettre de migrer et former une tumeur secondaire. Il est
cependant maintenant évident que les processus biologiques mis en place par l'hôte jouent un
rôle important dans ce processus. Des facteurs tels que l'interaction entre la tumeur et son micro-
environnement au sein de l'hôte, la surveillance immunitaire de la tumeur ainsi que la
configuration du système vasculaire impactent la croissance tumorale, la prise d'origine à des
sites distants et la spécificité organique de métastases. L'immunoediting du cancer contribue à la
résistance de l'hôte et fournit une pression sélective qui détermine l'issue de la maladie. En plus,
le créneau métastatique sécrète des effecteurs qui le préparent pour la colonisation des cellules
cancéreuses et la croissance métastatique. Afin d’identifier de nouveaux gènes de l’hôte
contrôlant le processus de métastase, notre laboratoire a mis en place des plates-formes de
génétique classique et inverse (forward and reverse genetics). Avec un criblage de souris ayant
subis une mutagénèse aléatoire avec le produit chimique (N-ethyl-N-nitrosourea [ENU] germline
mutagenesis), nous avons identifié deux familles de souris résistantes à la métastase. Le
séquençage génomique des exons a révélé une liste de gènes mutés candidats, incluant Nbeal1,
Cadm3, Ap1g2, qui pourraient être à la cause de cette résistance. L’identification de la mutation à
l’origine de ces phénotypes déviants ainsi que leur fonction dans le processus de métastase
permettra une meilleure compréhension de la pathogenèse du cancer et des métastases, et
pourrait révéler de nouvelles cibles pour améliorer les traitements du cancer.

4
TABLE OF CONTENTS
Abstract ...........................................................................................................................................2
Résumé ............................................................................................................................................3
Contributions of Authors ..............................................................................................................6
Acknowledgments ..........................................................................................................................7
List of Abbreviations .....................................................................................................................8
Literature Review ........................................................................................................................12
1. Colorectal Cancer ............................................................................................................12
1.1 CRC Epidemiology...........................................................................................................12
1.2 Intestinal Homeostasis ......................................................................................................14
1.2.1 Characteristics of NOD-Like receptors……………………………………………16
1.3 CRC Disease Initiation and Progression ..........................................................................20
1.4 CRC Genome-wide Association Studies ..........................................................................23
1.5 WNT Signalling in CRC ...................................................................................................28
1.6. Genetic Instability in CRC ..............................................................................................29
1.7 Familial CRC ....................................................................................................................31
2. Hallmark of Cancer .........................................................................................................36
2.1 Characteristics of Cancer Cells ........................................................................................36
3. CRC Microenvironment ..................................................................................................39
3.1 Intestinal Microenvironment ............................................................................................39
3.2 CRC Stem Cell Niche .......................................................................................................40
3.3 CRC stroma ......................................................................................................................41
3.4 Immune Cell Involvement in CRC ...................................................................................44
3.5 CRC Vasculature ..............................................................................................................46
4. Pre-Metastatic Niche and Organotropism in CRC .......................................................49
4.1 CRC Metastasis ................................................................................................................53
5. Cancer Immunoediting ....................................................................................................55
5.1 Elimination .......................................................................................................................55
5.2 Equilibrium .......................................................................................................................56
5.3 Escape ...............................................................................................................................60
6. Discovery of Host Genetic Determinants of CRC .........................................................62
6.1 Genetic Screening and Candidate Genes ..........................................................................62
6.2 ENU Mutagenesis .............................................................................................................63
Goals of the Study ........................................................................................................................66

5
Materials and Methods ................................................................................................................67
1. Mice ...................................................................................................................................67
2. Generation of ENU Mutants ..............................................................................................67
3. Model of CRC Lung Metastasis ........................................................................................68
4. MC38met-Luc Cell Culture ...............................................................................................68
5. ENU Screen .......................................................................................................................69
6. DNA Extraction and Purification .......................................................................................69
7. Exome Sequencing.............................................................................................................70
8. Antibody Depletion ............................................................................................................71
9. Lung Digestion/Cell Isolation ...........................................................................................71
10. Flow Cytometry ................................................................................................................71
11. Bioluminescence Imaging ..................................................................................................72
12. Histopathology ...................................................................................................................72
Results ...........................................................................................................................................74
1. Candidate Genes ................................................................................................................74
2. ENU Screen for Host Genetic Determinants of CRC Metastasis .................................................. 77
Discussion .....................................................................................................................................84
Conclusion ……………………………………………………………………………………. 89
Bibliography .................................................................................................................................92
Figures and Tables .....................................................................................................................117
1. Figure 11 ..........................................................................................................................122
2. Figure 12 ..........................................................................................................................123
3. Figure 13 ..........................................................................................................................124
4. Figure 14 ..........................................................................................................................125
5. Figure 15 ..........................................................................................................................126
6. Figure 16 ..........................................................................................................................127
7. Figure 17 ..........................................................................................................................127
8. Figure 18 ..........................................................................................................................128
9. Figure 19 ..........................................................................................................................128
10. Figure 20 ..........................................................................................................................129
11. Figure 21 ..........................................................................................................................129
12. Table 2 .............................................................................................................................130
13. Table 3 .............................................................................................................................131
14. Table 4 .............................................................................................................................132

6
CONTRIBUTIONS OF AUTHORS
This thesis was written solely by myself, and was edited by Dr. Maya Saleh. Parts of the
introduction are derived from a review article written by myself and Yifei Zhong, “Functions of
NOD-Like Receptors in Human Diseases” published October 16, 2013 in Frontiers in
Immunology 4: 333. Mice were injected with the help of Patricia D’Arcy and monitored with the
help of Joshua Rinz. Lung cell isolation and Flow cytometry experiments were completed with
the help of Phoebe Zhong and Dr. Alexandre Morizot. Data analysis was completed with the
help of Dr. Alexandre Morizot, Dr. Ian Gael Rodrigue-Gervais and Dr. Maya Saleh.

7
ACKNOWLEDGEMENTS
I would like above all to thank my supervisor, Dr. Maya Saleh for giving me the opportunity to
work on this project. Her guidance and support have been instrumental in my work, and have had
a formative influence on my work in her lab.
I would also like to acknowledge the numerous members of Maya’s lab who have helped me
over the course of my M.Sc., with special thanks to Josh Rinz, Dr. Alexandre Morizot, Dr. Ian
Gael Rodrigue-Gervais, Phoebe Zhong, Claudia Champagne and Maryse Dagenais for their
contributions to my project.
Other individuals who have made my project possible; thank you to Dr. Silvia Vidal and Dr.
Phillipe Gros for providing mice, Patricia D’Arcy for helping me with mouse tail i.v. injections,
Gabriel Leiva for his help with DNA extraction and exome sequencing analysis, as well as my
committee members, Dr. Ciriaco Piccirillo and Dr. Woong-Kyung Suh for their guidance and
advice.

8
LIST OF ABBREVIATIONS
ACK Ammonium-Chloride-Potassium
AFAP Attenuated Familial Adenopolyposis
AMP Antimicorbial peptide
ANNOVAR Annotate Variation Software
AOM Azoxymethane
AP-1 Activator-protein 1
AP1G2 Adaptor-related protein complex 1 gamma 2 subunit
APC Adenomatous polyposis coli
APF Australian Phenomics Facility
ARIDIA AT-rich interactive domain 1A
BCL2 B-cell lymphoma 2
BER Base excision repair
BID BH3 interacting domain
BIRC3 Baculoviral IAP repeat-containing protein 3
BMP Bone morphogenic protein
BMPR1A Bone morphogenic protein receptor type 1A
CA 9-19 Cancer antigen 9-19
CA4P Combrestatin A-4 phosphate
CACNA1G Calcium channel voltage-dependent T-type alpha 1G
subunit
CADM3 Cell adhesion molecule 3
CAF Cancer-associated Fibroblast
CARD Caspase activation and recruitment domain
CASP1,12 Caspase-1, 12
CCL2 Chemokine (C-C motif) ligand 2
CCND2 Cyclin D2
CCRK Cell cycle-related kinase
CDK4,6 Cyclin-dependent kinase 4
CEA Carcinoembryonic antigen
cIAP1/2 Cellular inhibitor of apoptosis 1/2
CIITA Class II major histocompatibility complex transactivator
CIMP CpG island methylator phenotype
CIN Chromosomal instability
CK1α Casein kinase 1 alpha
COX2 Cyclooxegenase 2
CRC Colorectal cancer
CSC Cancer stem cell
CSF Colony-stimulating factor
CTLA-4 Cytotoxic T-lymphocyte Antigen 4
CTNNB1 Catenin Beta 1
CXCR4 C-X-C receptor type 4
DC Dendritic cell
DNA Deoxyribonucleic acid

9
DSH Dishevelled
EDTA Ethylenediaminetetraacetic acid
EGF Epidermal growth factor
EGFR Epidermal growth factor receptor
ENU N-ethyl-N-nitrosourea
EPCAM Epithelial cell adhesion molecule
ERAS ES cell-expressed RAS
ERBB2 v-erb-b2 avian erythroblastic leukemia viral oncogene
homolog 2
ERK Extracellular signal-regulated kinase
EtBr Ethidium bromide
FAP Familial adenomatous polyposis
FBS Fetal Bovine Serum
FGF Fibroblast growth factor
FOXP3 Forkhead box p3
FZD Frizzled
G-CSF Granulocyte colony-stimulating factor
GJP Gastric juvenile polyposis
GM-1 Mono-sialo-tetra-hexosyl-ganglioside
GSK3 Glycogen synthase kinase 3
GTP Guanine 5’-triphosphate
GWAS Genome-wide association study
H&E Hemotoxylin and eosin
HEPES N-2-Hydroxyethylpiperazine-N-2-ethansulfonic acid
HGF Hepatocyte growth factor
HIF-1α Hypoxia-inducible factor 1 alpha
HNF4A Hepatocyte nuclear factor 4 alpha
HNPCC Hereditary non-polyposis colorectal cancer
i.p. Intraperitoneal
i.v. Intravenous
IBD Inflammatory bowel disease
IDO Indoleamine 2,3-dioxygenase
IEC Intestinal epithelial cell
IFN Interferon
Ig Immunoglobulin
IGF2 Insulin-like growth factor 2
IKKβ Inhibitor of NF-κB kinase subunit beta
IL Interleukin
ILC Innate lymphoid cell
IRF Interferon regulatory factor
ITCH Itchy E3 Ubiquitin protein ligase
JAK Janus Kinase
JNK c-Jun N-terminal kinase
JPS Juvenile polyposis syndrome
KRAS V-Ki-ras2-Kirsten rat sarcoma viral oncogene homolog
LDH-5 Lactate dehydrogenase 5

10
LEF Lymphoid enhancer-binding factor
LGR5 Leucine-rich repeat containing G-protein-coupled receptor 5
LHX1 LIM homeobox 1
LOX Lysyl oxidase
LRP5/6 Low density lipoprotein receptor-related protein 5/6
LRR Leucine-rich repeat
LUBAC Linear ubiquitin assembly complex
LY6G Lymphocyte antigen 6G
MAP MYH-associated polyposis
MAPK Mitogen-activated protein kinase
MCA Methylcholanthrene
MCP-1 Monocyte chemotactic protein 1
M-CSF Macrophage colony-stimulating factor
MDSC Myeloid-derived suppressor cell
MHC Major histocompatibility complex
MLH1/2/3 MutL homolog 1/2/3
MMP Matrix metalloproteinase
MMR Mismatch repair
MSH6/3 MutS homolog 6/3
MSI Microsatellite instability
mTOR Mammalian target of rapamycin
MYC c-Myc
MYH MutY homolog
NABP1 Nucleic acid binding protein 1
NAV2 Neuron navigator 2
NBEAL1 Neurobeachin-like 1
NEMO NF-κB essential modulator
NEUROG1 Neurogenin-1
NF-κB Nuclear factor kappa-light-chain-enhancer of activated B
cells
NK Natural Killer
NLR NOD-like receptor
NOD Nucleotide-binding oligomerization domain
PBS Phosphate-buffered saline
PCI Phenol-chloroform-isoamyl
PD-1 Programmed death 1
PDGF-B Platelet-derived growth factor B
PD-L1 Programmed death-ligand 1
PI3K Phosphotidylinositol-4,5-bisphosphate 3 kinase
PIGF Placental growth factor
PJS Peutz Jeghers syndrome
PMS1/2 Post-meiotic segregation increased 1/2
PP2A Protein phosphatase 2A
PTEN Phosphatase and tensin homolog
PTPRκ Protein tyrosine phosphatase receptor type K
RAG1/2 Recombination-activating gene 1/2

11
RAS Rat sarcoma
REG3γ Regenerating islet-derived protein 3 gamma
RIPK2/3 Receptor-interacting serine-threonine kinase 2/3
ROS Reactive oxygen species
RSPO2/3 Roof plate-specific spondin-2/3
RTK Receptor tyrosine kinase
RUNX3 Runt-related transcription factor 3
SAMtools Sequence Alignment/Map tools software
SDF-1α Stromal cell-derived factor 1 alpha
SHH Sonic hedgehog
SMAD SMA/MAD Homology
SNP Single nucleotide polymorphism
SNV Single nucleotide variant
SOCS1 Suppressor of cytokine signalling 1
SOX9 Sex-determining region box 9
STAT Signal transducers and activators of transcription
STK11 Serine/threonine kinase 11/24
TAB2/3 TAK1-binding protein
TAG-72 Tumor-associated glycoprotein
TAK1 Transforming growth factor β activated kinase-1
TAM Tumor-associated macrophage
TCF7L1 Transcription factor 7
TCRβ T cell receptor beta chain
TDO Tryptophan 2,3-dioxygenase
TDSF Tumor-derived secreted factor
TE Tris-EDTA
TGFβ/α Transforming growth factor beta
TLR Toll-like receptor
TP53 Tumor protein p53
TRAF Tumor necrosis factor associated factor
TRAIL Tumor necrosis factor apoptosis-inducing ligand
VDA Vascular disrupting agent
VEGF Vascular endothelial growth factor
VLA-4 Very late antigen-4
WT Wild type
XIAP X-linked inhibitor of apoptosis

12
LITERATURE REVIEW
1. Colorectal Cancer
1.1 CRC Epidemiology
Colorectal cancer (CRC) is the third most common malignancy in humans and the fourth most
common cause of cancer-related deaths, with over 1.2 million cases occurring globally each year,
and accounting for 600, 000 deaths in these patients (Ferlay et al., 2010). The Canadian cancer
society is predicting for 2014 alone, that 24,400 Canadians will be diagnosed with CRC, while
9,300 will die from the disease, accounting for 13% of 2014 cancer cases and 12% of all cancer
deaths in Canada respectively (Canadian Cancer Society Statistics 2014). The disease has a
markedly higher incidence in men, with 13, 500 Canadian men expected to be diagnosed in
2014, as opposed to 10,800 Canadian women expected to develop the disease (Canadian Cancer
Society Statistics 2014).
The etiology of CRC is complex, with contributions from both extrinsic and intrinsic
factors such as gender, age, gut microbial composition, diet, smoking, and physical activity
(Colditz et al., 2000;Fedirko et al., 2011;Boyle et al., 2012;Hansen et al., 2013;Stegeman et al.,
2013). In the context of CRC, these factors ultimately combine to activate proto-oncogenes, such
as the WNT pathway transcription factor, CTNNB1, and repress tumor suppressors, such as the
“guardian of the genome” TP53, by directly mutating DNA or regulating gene activity through
epigenetics, such as the silencing of the mismatch repair enzyme MLH1 by hypermethylation
(Hammoud et al., 2013).
Environment plays a large role in susceptibility to CRC, with factors such as food-borne
mutagens, intestinal commensals and pathogens and chronic intestinal inflammation, such as that

13
occurring during inflammatory bowel diseases (IBD), conferring an odds ratios of 1.1-1.85, 3.5
and 1.5 for developing CRC, respectively (Gilsing et al., 2012;Kato et al., 2013;Toriola et al.,
2013). While CRC remains a common cancer in the developed world, factors such as the
Westernization of diet is increasing the previously low incidence of CRC in many developing
countries. With the prevalence of obesity and the metabolic syndrome in these countries, and the
recently acquired knowledge of the effect of diet on gut microbiota, establishing the link between
diet, microbiota, CRC development and progression will become one of the primary goals of
CRC research in years to come.
Diagnosis of CRC is most commonly done via endoscopy (Lieberman et al., 2012), a
procedure which allows physicians, with the aid of a thin tool known as a colonoscope, to
examine and view the inside of the colon for ulcers, polyps, tumors, and abnormal inflammation
or bleeding. During this procedure, clinicians may take biopsies of unusual growths in the colon
if CRC is suspected. These tissue samples, as well as samples of body fluids such as urine and
blood, can be used to confirm diagnosis, by testing for the presence of CRC tumor markers,
molecules which are generally produced and/or upregulated by transformed cells, or healthy cells
in close proximity to the tumor (Duffy et al., 2014). For example, the monitoring of
carcinoembryonic antigen (CEA) and tumor-associated glycoprotein 72 (TAG-72) protein
expression (Grizzle et al., 2001;Swiderska et al., 2014) have both been used to effectively
diagnose CRC cases in patients. Furthermore, analysis of the expression of prognostic factors
such CEA and carbohydrate antigens 9-19 (CA 9-19) can aid in staging of the disease, and in
determining the most effective therapeutic strategy for each patient (Huh et al., 2010;Peng et al.,
2013;Duffy et al., 2014;Swiderska et al., 2014).

14
1.2 Intestinal Homeostasis
The intestinal epithelial barrier is a highly organized organ which acts to segregate the entry of
microbes from the gut lumen into the lamina propria. The barrier itself consists of a single layer
of intestinal epithelial cells (IECs) covered by a stratified layer of mucous. While the outer layer
of mucous is colonized by commensal bacteria, the inner layer contains proteins such as
immunoglobulin A (IgA) and anti-microbial peptides (AMPs), which keep it largely void of
bacteria and thus provides a primary defence against potentially pathogenic organisms
(Johansson et al., 2008). The epithelium forms structures called villi, which protrude into the
lumen of the intestine. Microvilli cover these villi, increasing the surface area through which
nutrients can be absorbed to approximately 400m2 (Peterson and Artis, 2014). There are five IEC
subtypes, which are derived from the epithelial stem cells which are continuously proliferating at
the base of the villi, in the crypts. Following the production of daughter cells, instructive signals
subsequently direct the cells to move further up the crypt, differentiate, localize to specific
positions in the epithelium depending on cell function (Crosnier et al., 2006;van der Flier and
Clevers, 2009). Intestinal enterocytes are the most numerous within the epithelium, and generally
act to conserve barrier function and maintain tight junctions. Other IECs, such as goblet cells,
enteroendocrine cells and Paneth cells, act to produce mucous, hormones and AMPs,
respectively, while M cells continuously sample the luminal contents, presenting their findings to
nearby immune cells (Kim and Ho, 2010;Bevins and Salzman, 2011;Gallo and Hooper,
2012;Mabbott et al., 2013) (Figure 1).

15
Much energy is invested in maintaining homeostasis in the gut, ensuring that symbiotic
commensals are allowed to thrive, while preventing overgrowth or the crossing of potential
pathogens into the lamina propria (Garrett et al., 2010;Renz et al., 2012). In addition to
maintaining a mucous layer, the epithelium regenerates itself constantly, instructing cells at the
top of the villi to die and slough off, while maintaining constant stem cell proliferation in the
intestinal crypts. As a result, the intestinal epithelium renews itself every 2-3 days throughout the
human lifetime (Crosnier et al., 2006). lamina propria phagocytes survey the intestinal
Figure 1. The intestinal epithelial barrier consists of a highly organized mucosal surface
that prevents the entry of microbes into the lamina propria. The epithelium is constituted
of a single layer of intestinal epithelial cells (IECs) covered by a stratified mucus layer. The
five IEC lineages include enterocytes, mucus-producing goblet cells, hormone-producing
enteroendocrine cells, AMP-producing Paneth cells at the base of the crypts and finally, M
cells that sample antigens from the intestinal lumen in order to present them to nearby immune
cells. A high number of T cells, macrophages, IgA secreting B and plasma cells are present in
the lamina propria and the Peyer’s patches. (Adapted with permission by Frontiers Media:
Muniz L.R. et. al, Intestinal antimicrobial peptides during homeostasis, infection and disease.
Frontiers in Immunology 3, 310 (2012)).

16
environment, contributing to tissue repair and defence (Pull et al., 2005;Smythies et al., 2005),
while the presence of commensals in the lumen can stimulate resident dendritic cells (DCs) to
secrete IL-12, activating interferon-γ (IFNγ) secretion by Th1 cells. Th17-secreted cytokines
such as IL-17 can recruit neutrophils and initiate acute inflammation, while Th17-secreted IL-22
can help repair the epithelial barrier and stimulate AMP secretion by IECs. To avoid unnecessary
overt inflammation, retinoic acid produced by CD103+ DCs triggers the induction of FOXP3+
regulatory T cells (Tregs), which dampen the actions of effector T cells through the secretion of
IL-10 and/or TGFβ (Asseman et al., 1999;Johansson-Lindbom et al., 2005;Li et al., 2007).
Importantly, IECs themselves are equipped with surface and cytosolic receptors which
recognize microbial-associated molecular patterns, such as bacterial flagellin, and danger-
associated molecular patterns, such as environment-derived toxins. Members of the surface-
expressed toll-like receptor (TLR) family, for instance, can recognize extracellular microbial
components, and trigger the release of AMPs such as REG3γ in response (Brandl et al., 2007).
Members of the cytosolic NOD-like receptor (NLR) family, can similarly sense and respond to
intracellular microbial or danger signals, leading to a cascade of events that result in the release
of IL-1β and IL-18 among other inflammatory signals.
1.2.1 Characteristics of NOD-like Receptors
The characteristic feature of NLRs is a central NOD (or NACHT) domain, required for
oligomerization, an N-terminal homotypic protein-protein interaction domain and a C-terminal
series of leucine-rich repeats (LRRs) involved in agonist sensing or ligand binding (Figure 2a).
Upon ligand binding, the auto-inhibitory LRR undergoes a conformational change, which
exposes the N-terminal domain allowing interaction with downstream signaling adaptors or
effectors and formation of an oligomeric complex (Inohara et al., 1999;Said-Sadier and Ojcius,

17
2012). NLR platforms that recruit and activate the inflammatory protease caspase-1 are referred
to as inflammasomes. Caspase-1 is required for the processing and maturation of the
inflammatory cytokines IL-1β and IL-18 and the induction of an inflammatory form of cell death
termed pyroptosis (Han et al., 2001;Willingham et al., 2009). While most NLRs, including the
highly-studied NLRP3, have been reported to exert their effects via the inflammasome, other
NLRs, such as NOD1, NOD2, NLRP10, NLRX1, NLRC5 and CIITA do not directly engage the
inflammatory caspases, but instead activate nuclear factor-κB (NF-κB), mitogen-activated
protein kinases (MAPK) and interferon (IFN) regulatory factors (IRF) to stimulate innate
immunity (Figure 2b).
2A.

18
Activation of NOD1 and NOD2 occurs differently from the inflammasome-forming
NLRs, and follows the cytosolic recognition of peptidoglycan ligands that triggers
oligomerization of the receptors via their NOD domain and the recruitment of mediators needed
to form a signaling complex referred to as the nodosome (Tattoli et al., 2007). The nodosome is
directed to the point of bacterial entry on the plasma membrane of polarized epithelial cells by
the regulatory protein FRMBP2 (Lipinski et al., 2012). NOD1 and NOD2 both interact with
2B.
Figure 2. NLR structure and pathways. 2A) With the capacity to sense a
wide range of MAMPs and DAMPs, inflammasome-forming NLRs can
assemble into a macromolecular complex to activate caspase-1. 2B) Non-
inflammasome-forming NLRs, such as NOD1 and NOD2 can respond to
cytosolic bacterial peptides and genomic material, leading to the recruitment
of adaptors which can activate potent immune effectors such as NF-κB and
IRF3. (Bottom panel adapted with permission by Frontiers Media: Kinio A.
& Zhong Y., Functions of NOD-Like Receptors in Human Diseases,
Frontiers in Immunology 4, 333 (2013))

19
RIPK2, via a CARD-CARD homotypic interaction (Kobayashi et al., 2002;Lecine et al.,
2007;Park et al., 2007;Nembrini et al., 2009). This association results in the recruitment of a
number of E3 ubiquitin ligases, including TNF receptor-associated factors (TRAFs) (Hasegawa
et al., 2008), cellular inhibitor of apoptosis (cIAP)1 and cIAP2 (Bertrand et al., 2009), X-linked
inhibitor of apoptosis (XIAP) (Krieg et al., 2009;Damgaard et al., 2012) and ITCH (Tao et al.,
2009). K63-linked ubiquitination of RIPK2 has been established as a means to construct protein
scaffolds that transduce downstream signaling. In a step-wise fashion, ubiquitination of RIPK2
leads to activation and recruitment of the TAK1 complex, consisting of TAK1 in association
with TAK1-binding protein (TAB)2 and TAB3. The kinase activity of TAK1 leads to
phosphorylation events that activate AP-1 and NF-κB. In parallel to cIAP-induced ubiquitination
of RIPK2, XIAP’s enzymatic activity results in the formation of polyubiquitin chains on RIPK2,
which serve as a platform to engage another E3 ligase complex known as the Linear Ubiquitin
Assembly Complex (LUBAC) (Ikeda et al., 2011;Damgaard et al., 2012). LUBAC attaches
linear ubiquitin chains to the regulatory protein NEMO, allowing for activation of the IKK
complex. The kinase activity of IKKβ results in the phosphorylation and degradation of the
inhibitor of NF-κB (IκB), allowing for NF-κB dimers to translocate to the nucleus and induce
proinflammatory gene expression (Hasegawa et al., 2006). Besides activating NF-κB, NOD1 and
NOD2 have also been shown to activate the p38, JNK and ERK MAPK pathways (Pauleau and
Murray, 2003;Kobayashi et al., 2005;Park et al., 2007) and to interact with other NLRs such
NLRP1 and NLRP12 (Hsu et al., 2008;Wagner et al., 2009). In addition to it’s pro-apoptotic
function, the BH3-only protein BID has been implicated in NOD1 signalling to NFkB and MAP
Kinase pathways.

20
1.3 CRC Disease Initiation and Progression
In most individuals, the most initial indicator of future CRC occurs as an adenomatous polyp
(Dewanji et al., 2011). These benign growths often occur in the large intestine, and are generally
considered to be genetically stable, often remaining dormant for years before becoming clinically
relevant (Luebeck and Moolgavkar, 2002;Jones et al., 2008). Following a multi-step progression
model, genetic and epigenetic changes within adenomatous polyps can lead to changes in their
histological staging, determining whether the polyps become cancerous, or whether they remain
benign. For instance, a subclass of polyps is associated with a mutation in the oncogene, KRAS2,
leading to increased activity, but generally resulting in non-malignant intestinal growths (Nucci
et al., 1997). On the other hand, other lesions in the intestine may be the result of the inactivation
of the tumor suppressor gene, adenomatous polyposis coli (APC), whose inactivation increases
the likelihood of the lesion progressing to cancer, and whose inactivation has been observed in
80-90% of sporadic CRC cases (Ahearn et al., 2012). The loss of APC activity results in
disruption of the WNT/β-catenin signalling pathway, resulting in loss of cell cycle check-points,
increased cell division, and increased cell motility (Moon et al., 2014). Because of the strong
effects of the dysregulation of the WNT/β-catenin pathway, inhibition of APC activity, or
mutations in genes encoding other components within the pathway, such as β-catenin and Axin2,
are considered to be important to the initiation of sporadic CRC (Ahearn et al., 2012).
Following the disruption in WNT/β-catenin signalling, and resulting polyp formation,
subsequent mutations affecting KRAS activity are associated with adenoma progression and
poor prognosis (Janssen et al., 2006). Because KRAS acts as a regulator of extracellular signal-
regulated kinase (ERK) and phosphotidylinositol 3 kinase (PI3K) pathways, mutations fixing
KRAS in its GTP-bound active form result in constitutive activation of these kinases, influencing

21
cell survival, proliferation, metabolism and motility (Mendoza et al., 2011). The importance of
this step in CRC progression is reflected in the fact that approximately 40% of sporadic CRC
cases express activating KRAS mutations (Bos et al., 1987;Andreyev et al., 2001), and 5-22% of
patients without KRAS mutations express activating mutations in BRAF (Oliveira et al.,
2007;Roth et al., 2010;Zlobec et al., 2010), which similarly plays a role in cancer growth and
survival due to its role in regulating the MAPK signalling cascade.
As the polyp becomes larger and more aggressive, mutations in TGFβ signalling pathway
further allows for increased growth, differentiation and migration, but also promotes
angiogenesis and immune cell regulation, and thus often accompanies the transition from
adenoma to carcinoma. Disabling mutations in the proteins involved in this pathway, such as
TGFβ receptor II (TGFβRII), SMAD2 and SMAD4 are found in 30% (Biswas et al., 2008), 5%
(Fleming et al., 2013) and 10% of sporadic CRC cases, respectively (Koyama et al., 1999;Miyaki
et al., 1999;Iacobuzio-Donahue et al., 2004;Fleming et al., 2013).
Another gene of importance in the progression of adenoma to carcinoma and metastasis
is TP53, a gene which product, p53, prevents tumor formation by suppressing cell growth,
repairing the genome, or triggering cell death when genome damage is deemed too great (Balint
and Vousden, 2001). Inactivating mutations in TP53 thus have dire consequences for the
maintenance of genome stability and integrity, and, as a result, are observed in approximately
45% of all CRC cases (Petitjean et al., 2007) (Figure 3A).
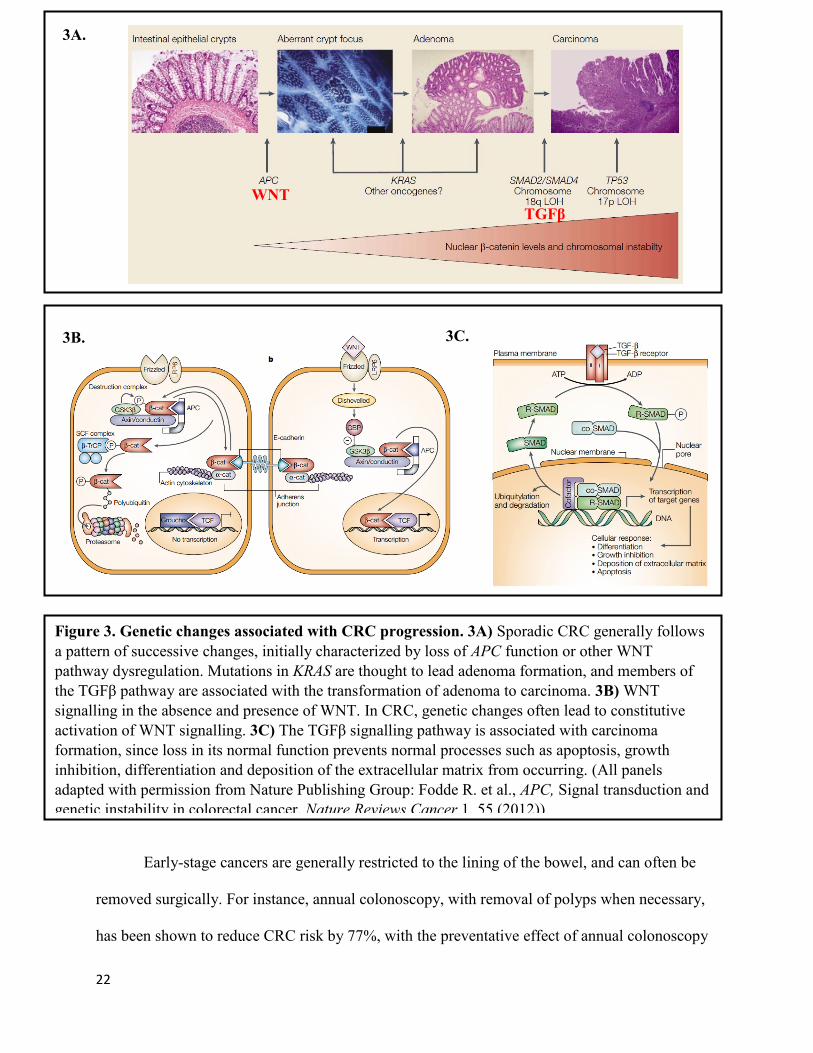
22
Early-stage cancers are generally restricted to the lining of the bowel, and can often be
removed surgically. For instance, annual colonoscopy, with removal of polyps when necessary,
has been shown to reduce CRC risk by 77%, with the preventative effect of annual colonoscopy
3A.
WNT
TGFβ
3C. 3B.
Figure 3. Genetic changes associated with CRC progression. 3A) Sporadic CRC generally follows
a pattern of successive changes, initially characterized by loss of APC function or other WNT
pathway dysregulation. Mutations in KRAS are thought to lead adenoma formation, and members of
the TGFβ pathway are associated with the transformation of adenoma to carcinoma. 3B) WNT
signalling in the absence and presence of WNT. In CRC, genetic changes often lead to constitutive
activation of WNT signalling. 3C) The TGFβ signalling pathway is associated with carcinoma
formation, since loss in its normal function prevents normal processes such as apoptosis, growth
inhibition, differentiation and deposition of the extracellular matrix from occurring. (All panels
adapted with permission from Nature Publishing Group: Fodde R. et al., APC, Signal transduction and
genetic instability in colorectal cancer, Nature Reviews Cancer,1, 55 (2012)).

23
increasing as individuals age (Brenner et al., 2011). With more advanced disease, surgery may be
combined with less frequently used methods of treatment, such as chemotherapies which fatally
damage the DNA of quickly proliferating cells, most commonly using the anti-metabolite 5-
Fluorouracil (Colorectal Cancer Association of Canada, 2011), or more targeted drugs such as
Cetuximab, a monoclonal antibody which targets the epidermal growth factor receptor (EGFR)
in patients with metastatic CRC. Radiation therapy is also common, and may be performed via
an external beam, or by placing radioactive pellets directly at the site of the tumor, as is done in
brachytherapy. Surgery provides the most effective option for CRC patients, however, more
invasive CRC may be treated with a combination of surgery, chemotherapy and radiation.
Unfortunately, invasive CRC is often fatal and treatment is usually palliative, focusing on
extending life and minimizing discomfort.
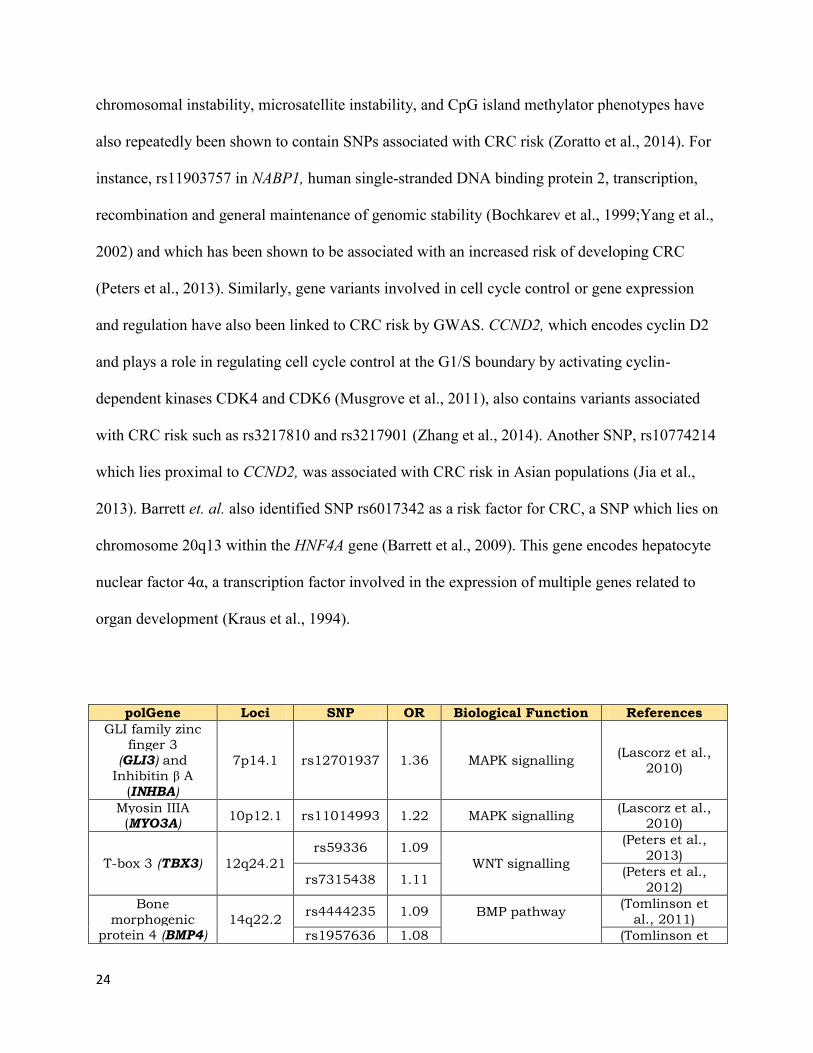
1.4 CRC Genome-wide Association Studies
In recent years, Genome-wide Association Studies (GWAS) have expanded the database of
genes and SNPs associated with CRC, identifying over 40 common genetic variants affecting the
risk of developing CRC(Table 1) (Zhang et al., 2014). Most of these variants play have minimal
effect on the risk of CRC and generally have an odds ratio (OR) of less than 1.2 (Zhang et al.,
2014). SNPs associated with increased risk of developing CRC affect signal transduction
pathways, such as the WNT/β-catenin signalling pathway or the TGFβ/BMP and MAPK
pathways. For example, the rs59336 risk allele was identified in TBX3, a downstream target of
WNT/β-catenin signalling (Tomlinson et al., 2008), and three SNPs in SMAD7, rs4939827,
rs12953717 and rs4464148 have been associated with an increased CRC risk (Broderick et al.,
2007;Tenesa et al., 2008). In addition, genes related to genomic instability mechanisms, such as
24
chromosomal instability, microsatellite instability, and CpG island methylator phenotypes have
also repeatedly been shown to contain SNPs associated with CRC risk (Zoratto et al., 2014). For
instance, rs11903757 in NABP1, human single-stranded DNA binding protein 2, transcription,
recombination and general maintenance of genomic stability (Bochkarev et al., 1999;Yang et al.,
2002) and which has been shown to be associated with an increased risk of developing CRC
(Peters et al., 2013). Similarly, gene variants involved in cell cycle control or gene expression
and regulation have also been linked to CRC risk by GWAS. CCND2, which encodes cyclin D2
and plays a role in regulating cell cycle control at the G1/S boundary by activating cyclin-
dependent kinases CDK4 and CDK6 (Musgrove et al., 2011), also contains variants associated
with CRC risk such as rs3217810 and rs3217901 (Zhang et al., 2014). Another SNP, rs10774214
which lies proximal to CCND2, was associated with CRC risk in Asian populations (Jia et al.,
2013). Barrett et. al. also identified SNP rs6017342 as a risk factor for CRC, a SNP which lies on
chromosome 20q13 within the HNF4A gene (Barrett et al., 2009). This gene encodes hepatocyte
nuclear factor 4α, a transcription factor involved in the expression of multiple genes related to
organ development (Kraus et al., 1994).
polGene Loci SNP OR Biological Function References
GLI family zinc
finger 3 (GLI3) and
Inhibitin β A
(INHBA)
7p14.1 rs12701937 1.36 MAPK signalling (Lascorz et al.,
2010)
Myosin IIIA (MYO3A)
10p12.1 rs11014993 1.22 MAPK signalling (Lascorz et al.,
2010)
T-box 3 (TBX3) 12q24.21
rs59336 1.09
WNT signalling
(Peters et al.,
2013)
rs7315438 1.11 (Peters et al.,
2012)
Bone
morphogenic protein 4 (BMP4)
14q22.2 rs4444235 1.09 BMP pathway
(Tomlinson et
al., 2011)
rs1957636 1.08 (Tomlinson et

25
al., 2011)
DAN family BMP
antagonist (GREM1)
15q13.3
rs16969681 n/a
BMP pathway
(Tomlinson et
al., 2011)
rs4779584 n/a (Tomlinson et
al., 2011)
rs11632715 n/a (Tomlinson et
al., 2011)
SMAD family
member 7
(SMAD7)
18q21
rs4939827 1.2
TGFβ and WNT
signalling
(Broderick et al., 2007;Tenesa et
al., 2008;Cui et
al., 2011)
rs12953717 1.17 (Broderick et al.,
2007)
rs4464148 1.15 (Broderick et al.,
2007)
rs4939827 1.12 (Tomlinson et
al., 2008;Peters
et al., 2013)
rs4939827 1.14 (Peters et al.,
2012)
Bone
Morphogenic
protein 2 (BMP2)
20p12.3
rs961235 1.12
BMP Pathway
(Tomlinson et
al., 2011)
rs4813802 1.09 (Tomlinson et
al., 2011)
Casein kinase 2, α 1 polypeptide
(CSNK2A1)
20p13 rs6038071 2.64 MAPK signalling (Lascorz et al.,
2010)
Nucleic acid
binding protein 1
(NABP1)
2q32.3 rs11903757 1.16 DNA maintenance
and repair
(Peters et al.,
2013)
Paired-like
homeodomain
(PITX1)
5q31.1 rs647161 1.11
RAS pathway,
activation of TP53,
telomerase activity
(Jia et al., 2013)
Cyclin-dependent
kinase inhibitor 1A (CDKN1A)
6p21 rs1321311 1.1
Microsatellite instability, DNA
repair, genomic
instability
(Dunlop et al.,
2012)
Polymerase DNA-directed δ3
(POLD3) 11q13.4 rs3824999 1.08 DNA MMR and BER
(Dunlop et al.,
2012)
Tumor protein
p53 (TP53)
17p13 rs78378222 1.39 Regulatore of cell
division
(Stacey et al.,
2011)
Laminin gamma 1 (LAMC1)
1q25.3 rs10911251 1.09 Gene transcription
(Peters et al.,
2013)
Dual-specificity
phosphatase (DUSP10)
1q41
rs6691170 1.06
Inactivates p38
(Houlston et al.,
2010)
rs6687758 1.09 (Houlston et al.,
2010)
Laminin β1 (LAMB1)
7q31 rs88
6774 1.17
Anchoring the single-
layered epithelium
(Barrett et al.,
2009)
POU class 2
associating factor 1 (POU2AF1)
11q23 rs3802842 1.1 Transcriptional
coactivator
(Tenesa et al.,
2008)

26
Cyclin D2 (CCND2)
12p13.32
rs10774214 1.09
Cell-cycle transition
(Jia et al., 2013)
rs3217810 1.2 (Peters et al.,
2013)
rs3217901 1.1 (Peters et al.,
2013)
Disco-interacting
protein 2 B (DIP2)
12q13.13 rs11169552 1.09 Cell morphogenesis (Houlston et al.,
2010)
E-cadherin (CDH1)
16q22 rs1728785 1.17
Epithelial restitution,
repair following
mucosal damage
(Barrett et al.,
2009)
Rho GTPase
binding protein 2 (RHPN2)
19q13.33 rs10411210 1.15 Actin cytoskeleton (Houlston et al.,
2008)
Large laminin A5 (LAMA5)
20q13.33 rs4925386 1.08 BMP pathway (Houlston et al.,
2010)
Shroom family
member 2 (SHROOM2)
Xp22.2 rs5934683 1.07 Cell morphogenesis (Dunlop et al.,
2012)
Eukaryotic
translation
initiation factor
3, subunit H (EIF3H)
8q23.3 rs16892766 1.25 Translation initiation (Tomlinson et
al., 2008)
POU class 5
homeobox 1B (POU5FIP1)
8q24 rs7014348 1.19 Transcriptional
activator (Tenesa et al.,
2008)
Activating
transcription factor 1 (ATF1)
12q13.13 rs7136702 1.06 Transcription (Houlston et al.,
2010)
Transcription
factor hepatocyte nuclear factor 4α
(HNF4A)
20q13.12 rs6017342 1.11 Transcription (Barrett et al.,
2009)
- 1p36.12 rs7524102 1.1 - (Barrett et al.,
2009)
Chromosome 1
open reading
frame 21 (Clorf21)
1q31 rs16823149 - - (Lascorz et al.,
2010)
Plasminogen-like
A, non-coding RNA (PLGLA)
2q12 rs4574118 - - (Lascorz et al.,
2010)
Myoneurn gene (MYNN)
3q26.2 rs10936599 1.08 Unknown (Houlston et al.,
2010)
Non-SMC condensing I
complex, subunit G (NCAPC)
4p15.3 rs41
40904 - -
(Lascorz et al.,
2010)
Organic cation
transporter (SLC22A3)
6q25.3 rs7758229 1.28
Transport of cationic
drugs, toxins, and
endogenous
metabolism
(Cui et al., 2011)
- 8q24 rs6983267 1.18 - (Cui et al., 2011)
27
rs7837328 1.17 (Cui et al., 2011)
Transducin-like
enhancer of spit
4 (TLE4)
9q21.3 rs2209907 - - (Lascorz et al.,
2010)
- 10p14 rs10795668 1.12 - (Tomlinson et
al., 2008)
- 13q13.3 rs95
48988 1.1 -
(Barrett et al.,
2009)
Phospholipase C-beta 1 (PLCB1)
20p12.3 rs2423279 1.1 Unknown (Jia et al., 2013)
- 3p21.31 rs8180040 1.28 -
(Fernandez-
Rozadilla et al.,
2013)
- 8p12 rs12548021 1.28 -
(Fernandez-
Rozadilla et al., 2013)
- 8q22.1 rs3104964 1.27 -
(Fernandez-
Rozadilla et al.,
2013)
- 5q21.3 rs367615 1.35 - (Jiao et al.,
2012)
- 7p15.3 rs39453 1.28 - (Jiao et al.,
2012)
- 4q13.2 rs17730929 1.47 - (Jiao et al.,
2012)
Tryptophan
Hydroxylase 2
(TPH2) 12q21.1 rs10879357 1.25 Catalyzes serotonin
(Jiao et al.,
2012)
- 9q22.32 rs10114408 1.37 - (Jiao et al.,
2012)
- 3p24.3 rs4591517 1.06 - (Jiao et al.,
2012)
Synaptojanin 2
(SYNJ2) 6q25.3 rs9365723 1.27
Inhibits clathrin-
mediated endocytosis
(Jiao et al.,
2012)
Chromosome 5
Open Reading
Frame 66
(C5Orf66)
5q31.1 rs647161-A
1.11
Unknown (Jia et al., 2013) 1.17
Serine/arginine-
rich splicing factor 10
pseudogene 2 (SRSF10P2)
20p12.3 rs2423279-
C 1.1 1.14
Unknown (Jia et al., 2013)
Heat shock 70
kDa protein 12A
(HSPA12A) 10q25.3 rs1665650 1.13
Stabilization of
proteins (Jia et al., 2013)
- 4q22.2 rs13130787 1.09 - (Peters et al.,
2013)
- 20p12.3 rs961253 1.12 - (Houlston et al.,
2008)
Cadherin 1, Type
1 E-cadherin 16q22.1 rs9929218 1.1
Calcium-dependant
cell-cell adhesion
(Houlston et al.,
2008)

28
(CDH1)
- 8q24.21 rs10505477 1.17 - (Zanke et al.,
2007)
1.5 WNT Signalling in CRC
The WNT signalling pathway is a highly conserved pathway vital for processes such as
embryogenesis, tissue homeostasis and cancer pathogenesis (Voloshanenko et al., 2013). The
canonical WNT pathway is vital for intestinal tissue renewal and intestinal stem cell regulation.
The process of stem cell division is one of the key processes that is disrupted during CRC. In a
healthy gut, WNT signalling is predominant at the base of the intestinal crypts, supporting
extensive proliferation, but diminishes towards the open end of the crypt, where pathways such
as TGFβ/BMP promote cell specialization, positioning and apoptosis (Reynolds et al., 2014).
Disruptions in the WNT signalling cascade can lead to aberrations in cell migration and division
and inappropriate epithelial-to-mesenchymal transitions. In the absence of Wnt, β-catenin is
phosphorylated by glycogen synthase kinase (GSK3) and casein kinase 1α (CK1α), which are
members of a destruction complex that also includes axin, adenomatosis polyposis coli (APC),
protein phosphatase 2A (PP2A) (He et al., 2004). Phosphorylation of β-catenin leads to its
ubiquitination, targeting its destruction in the proteosome (Peters et al., 1999;Sakanaka et al.,
1999;Amit et al., 2002;Liu et al., 2002). WNT binding to its receptor complex, comprised of the
proteins frizzled (Fz) and low-density-lipoprotein-related protein5/6 (LRP5/6), prevents β-
catenin degradation by disrupting the APC/Axin/GSK3 complex, recruiting it and the negative
regulator of signalling, Axin, to the cell membrane, where it binds to the cytoplasmic tail of
Table 1. GWAS-identified SNPs associated with risk of developing CRC

29
LRP5/6 (Bilic et al., 2007;Schwarz-Romond et al., 2007). Through unknown mechanisms, this
leads to the phosphorylation and activation of Dishevelled (Dsh), allowing β-catenin to
accumulate in the cytoplasm, translocate into the nucleus and induce a cellular response by
acting as a transcriptional co-activator. Alongside LEF/TCF transcription factors (Behrens et al.,
1996;Huber et al., 1996) , β-catenin can act as a transcriptional co-activator of a vast array of
target genes involved in CRC pathogenesis. These include genes such as the oncogene MYC,
CCND1, which encodes CyclinD1, and the prostaglandin-endoperoxide synthase, COX2 (Herbst
et al., 2014) (Figure 3B, C).
Misregulation of WNT signalling represents one of the earliest events in CRC, and is so
vital to CRC progression, that it is disrupted in >92% of sporadic CRC tumors (2012). Of these
cases, approximately 80% carry inactivating mutations in APC and 5% show activating
mutations in β-catenin (Morin et al., 1997; 2012). Recently, a group used RNA-seq data to
compare 70 primary colon tumors, identifying recurrent gene fusions of R-spondins, specifically
RSPO2 and RSPO3, in 10% of samples (Seshagiri et al., 2012). These proteins can act as
activation ligands on LRP6 and LGR5 and can further crosslink with WNT and FZD, as well as
inhibits the degradation of LRP6 and FZD receptors, facilitating WNT signalling (Jin and Yoon,
2012). These mutations generally occurred in samples lacking APC mutations, and the authors
were able to verify their ability to potentiate WNT signalling by expressing R-spondin fusion
constructs in HEK 293 T cells with a luciferase reporter for WNT signalling (Seshagiri et al.,
2012).
1.6 Genetic Instability in CRC

30
Transformation of the normal gut mucosa follows a series of events which gradually converts
healthy tissue into a carcinoma. The basis of this process lies in the inherent genomic instability
of cancer cells. This instability results in several distinct mutations which can activate oncogenes
and deactivate tumor suppressors to drive tumorigenesis. To date, three pathways are recognized
to be involved in this process: the Chromosomal Instability (CIN) pathway, the Microsatellite
Instability (MSI) pathway, and the CpG Island Methylator Phenotype (CIMP) pathway.
65-70% of sporadic CRC has been attributed to chromosomal instability (Mouradov et
al., 2013). The hallmark of chromosomal instability is the loss of whole, or large regions of,
chromosomes, resulting from errors in chromosome segregation during mitosis or in errors in
DNA repair mechanisms. These defects lead to aneuploidy, loss of heterozygosity and genomic
amplifications. In CRC, chromosomal instability often causes mutations in APC and KRAS.
Microsatellite instability occurs due to errors in DNA mismatch repair (MMR), and
occurs in approximately 15% of CRC patients (Kanth et al., 2014). Microsatellites are short,
repeating sequences of DNA found throughout the genome. While DNA MMR functions to
prevent errors in base insertion, base deletion and mis-matching of bases, the repetitive nature of
microsatellites renders them susceptible to errors during DNA replication. Silencing of the MMR
system, or components of the MMR system, such as MLH1, MSH2, MSH6, PMS2, MLH3,
MSH3, PMS1, or Exol is commonly seen in sporadic CRC via hypermethylation . Genome
analysis of 276 CRC samples by the Cancer Genome Atlas Network found 24 genes significantly
mutated within high MSI samples, including expected genes such as APC, KRAS, TP53 and
SMAD4, but also revealing new hits in ARID1A, SOX9 and FAM123B, all directly or indirectly
involved in WNT signalling, as well as genes which had changes in mRNA copy number, such
as ERBB2, involved in RTK/RAS signalling and IGF2, a component in the PI3K signalling

31
cascade. This analysis also revealed previously unreported chromosomal translocations, such as
the fusion of NAV2, which is involved in cell growth and migration, as well as TCF7L1, which is
downstream of WNT signalling (2012).
Along with DNA mutations, gene activity in CRC can be affected at the epigenetic level.
Epigenetics can alter the expression or activity of genes without changing the DNA sequence.
For example, DNA methylation, which frequently occurs at CpG dinucleotides can silence gene
expression. Changes in DNA methylation have been observed in CRC, often affecting the
expression of tumor suppressors such as APC, MCC and MLH1(Desai and Barkel, 2008).
Advanced age and lifestyle factors, such as diet and smoking, are associated with DNA
hypermethylation (Toyota et al., 1999;Samowitz et al., 2006). The term CIMP specifically refers
to the hypermethylation of at least three of five genes which have been selected as markers for
CIMP. These are SOCS1, NEUROG1, RUNX3, CACNA1G and IGF2 (Weisenberger et al.,
2006). CIMP-positive CRC accounts for approximately 15-20% of spontaneous CRC and has
distinct characteristics, particularly the tendency of CIMP positive tumors to harbour BRAF
mutations, microsatellite instability and poorly differentiated cells (Nosho et al., 2008).
1.7 Familial CRC
While the development of CRC is mainly attributed to environmental factors in most patients,
approximately 20% of CRC cases have a clear familial basis. Familial CRC syndromes are
linked to highly penetrant mutations in genes such as APC, BMPR1A, SMAD4 and STK11
(Aaltonen et al., 2007). Familial adenomatous polyposis (FAP) is one example of a highly
penetrant familial CRC syndrome, being caused by heritable, autosomal-dominant germline

32
mutations in the APC gene (Groden et al., 1991;Kinzler et al., 1991). Intermediate phenotypes
exist- for instance, patients with mutations in the 5’ end and exon 4 of APC can contain
anywhere from 2 to 500 polyps, while patients with exon 9 mutations generally register 1 to 150
adenomas and patients with a mutation in the 3’ end of APC presenting with fewer than 50
adenomas (Spirio et al., 1993;Brensinger et al., 1998;Pedemonte et al., 1998;Soravia et al.,
1998). However, there are as yet, no clear genotype-phenotype relationships established in
AFAP, likely indicating a role for modifier genes and highlighting a need to further investigate
the underlying factors which distinguish AFAP from classical FAP. AFAP affects 1 in 10,000
individuals and accounts for 4% of CRC cases (Bulow et al., 1996;Barnetson et al., 2006). The
non- ,or semi-functional presence of APC in these patients leads to the development of
adenomas, or pre-cancerous lesions, in the colon and rectum (Wasmuth et al., 2013;Aihara et al.,
2014). The dysregulation of the WNT/β-catenin pathway is often labelled as the “rate-limiting”
step in sporadic colorectal cancer, due to its ability to promote adenoma progression and initiate
genome instability. Therefore, the APC inactivating mutations inherited by FAP patients
inevitably leads to the development of CRC in patients by age 40 (Jasperson et al., 2010), a much
younger age than that for sporadic CRC, due to the removal of this initial threshold. Because of
the high risk of developing CRC, FAP patients generally undergo prophylactic surgery between
ages 15-25 years, to remove sections of the rectum and colon containing adenomas. This
treatment can reduce short-term CRC development, however, the effect is time-dependent, with a
42% incidence of neoplastic polyp formation in the ileal pouch 7 years after proctectomy (Wu et
al., 1998;Church, 2005;Kartheuser et al., 2006). Thus, endoscopic surveillance is of vital
importance in these patients, and should ideally be performed on an annual basis (Thompson-
Fawcett et al., 2001;Hurlstone et al., 2008).

33
In addition to classical FAP, an attenuated version, referred to as AFAP has been
described. Like classical FAP, AFAP originates from autosomal dominant mutations in APC.
However, patients present with <100 polyps, fewer colorectal adenomas, a lower lifetime cancer
risk, and generally delayed onset of polyp formation than patients diagnosed with FAP (Ibrahim
et al., 2014) . Similar to AFAP, MYH associated polyposis (MAP) also presents itself with <100
polyps and an increased risk of CRC development, but originates from recessive mutation in
MYH and is believed to affect 1-3% of CRC patients (Halford et al., 2003). MYH is found at
position 1p34 on chromosome 1, and belongs to a complex involved in DNA base excision repair
(Bolocan et al., 2011). The gastrointestinal tract is constantly subject to trauma from ingested
substances and infection with bacteria which induce DNA damage. For this reason, inactivating
mutations in MYH could prevent damaged DNA from being repaired and could thus facilitate
adenoma formation (Kim et al., 2004). First described in 2002, little is known about the etiology
and epidemiology of MAP, with diagnosis usually occurring concurrently with CRC diagnosis,
and treatment generally following the same guidelines as that for FAP and AFAP patients
(Bolocan et al., 2011).
Lynch syndrome (LS) is another hereditary CRC syndrome. It occurs because of
autosomal dominant mutations in one or several components of the DNA mismatch repair
system (MMR), such as MLH1, MSH2, MSH6 and PMS2. LS leads to 80% lifetime risk of
developing CRC and an increased risk of developing other cancers, such as ovarian or gastric
cancers (Sturgeon et al., 2013). Under circumstances where a familial CRC syndrome meets the
autosomal dominant inheritance criteria of LS, but no MMR mutations have been identified, the
syndrome is referred to as hereditary nonpolyposis colorectal cancer (HNPCC). The fact that 30-
50% of HNPCC cases are unexplained suggests that additional factors are implicated in disease

34
development. For example, two groups recently reported 3’ end deletions in the genomic region
of epithelial cell adhesion molecule (EPCAM) in 19% of tested HNPCC cases (Kovacs et al.,
2009;Ligtenberg et al., 2009). These deletions were upstream of MSH2 and correlated with
MSH2 protein loss, possibly due to epigenetic silencing, and genomic instability (Kovacs et al.,
2009). Lifetime CRC risk in EPCAM deletion carriers was estimated at 70%, similar to the risk
of individuals carrying mutations in MLH1 or MSH2 (Kempers et al., 2011). LS patients are
predisposed to develop stomach, pancreatic, ureter, renal, prostate, breast and liver cancers, and
female carriers may be at a higher risk of developing endometrial cancers than CRC
(Quehenberger et al., 2005). Despite the fact that LS patients are at risk for a variety of cancers,
colorectal screening remains the only effective surveillance procedure for LS patients at this
time, leading to a >50% decrease in CRC development and 65% decrease in mortality due to
CRC in patients (Jarvinen et al., 2000). Screening protocols designed to detect early cancers in
other organs in LS patients, such as the liver and ovaries, have had no impact on survival and the
complexity of treating multiple cancers in LS patients contributes to the difficulty healthcare
practitioners face in attempting to treat the disease. Treatment for LS-related CRC has been
controversial, with recommendations for more extensive surgery, despite decreased functional
outcome following surgery (Haanstra et al., 2012;Vasen et al., 2013). The recommendations
were provided following observations by two groups that the occurrence of secondary CRC
following partial colectomy remained at 16%, despite regular surveillance for 10 years (de Vos
tot Nederveen Cappel et al., 2002;Parry et al., 2011). However, LS patients can minimize their
risk of developing CRC by maintaining a healthy body weight (Botma et al., 2010;Win et al.,
2011), refraining from smoking (Diergaarde et al., 2007;Pande et al., 2010;Winkels et al., 2012)
and taking aspirin daily (Burn et al., 2011;Rothwell et al., 2011).

35
Peutz-Jeghers syndrome (PJS) is another familial CRC syndrome associated with
autosomal dominant mutations in the serine threonine STK11 gene on chromosome 19p13
(Hemminki et al., 1998;Jenne et al., 1998;Hosogi et al., 2008). This kinase plays a complex role,
acting as a regulator of cellular proliferation, through G1 cell cycle checkpoints and interaction
with the cyclin-dependent kinase inhibitor p21, induction of p53-dependent apoptosis (Tiainen et
al., 1999;Karuman et al., 2001;Tiainen et al., 2002), modulation of the WNT pathway (Lin-Marq
et al., 2005) and regulation of cell polarity and metabolism (Morton et al., 1992). Importantly,
STK11 also indirectly acts as a regulator of the mammalian target of rapamycin (mTOR)
pathway (Corradetti et al., 2004), which is also dysregulated in juvenile polyposis syndrome
(JPS) due to mutations in PTEN, BMPR1A and SMAD4. Like other familial CRC syndromes,
PJS results in the development of polyps in the gastrointestinal tract, as well as other sites such
as in the bronchi, bladder or gallbladder (Vogel et al., 2000). In addition, approximately 95% of
PJS patients exhibit mucocutaneous pigmented lesions, which may arise during infancy and
occur on areas such as fingers and toes, as well as in the mouth and nostril area (Beggs et al.,
2010). Because of the early onset of polyps, CRC can occur at a relatively early age. PJS patients
have a 57% lifetime chance of developing gastrointestinal cancers and an 85% risk of developing
any cancer, including pancreatic, gynaecological and breast (45% risk in females) cancers
(Hearle et al., 2006). Given the high chance of CRC and breast cancer occurrence, intensive
colorectal and breast surveillance is generally advocated, although the lack of evidence makes it
unclear whether these measures can increase survival (Beggs et al., 2010;Latchford et al., 2011).
Juvenile polyposis syndrome (JPS) is exceedingly rare and leads to the development of
colorectal polyps in young children with a family history of JPS, leaving them at a 39% lifetime
CRC risk (Brosens et al., 2011). Approximately 50-60% of the time the disease is attributed to

36
autosomal dominant mutations in SMAD4 and BMPR1A (Aretz et al., 2007;van Hattem et al.,
2008) ,both of which are involved in the BMP/TGFβ signalling pathway. A particularly
aggressive form of JPS is seen in patients with mutations in the tumor suppressor PTEN, a
tyrosine phosphatase mutated in prostate, breast and brain cancers (Li and Sun, 1997;Li et al.,
1997;Steck et al., 1997). JPS generally presents in one of two forms; the first, called juvenile
polyposis of infancy, leads to the development of polyps in the stomach, bowel and colon,
usually before the age of 2 years. Patients do not usually survive past an early age, and suffer
from symptoms such as diarrhea, haemorrhage and malnutrition (Brosens et al., 2011). Deletion
of BMPR1A or PTEN, both located on chromosome 10 are believed to be responsible for this
aggressive manifestation of JPS (Delnatte et al., 2006) . Generalized juvenile polyposis (GJP), in
which 50% of cases contain heterozygous germline mutations in SMAD4 or BMPR1A represents
less aggressive manifestations of the disease, with polyps presenting in late childhood or adult
life (Delnatte et al., 2006).
2. Hallmarks of Cancer
2.1 Characteristics of Cancer Cells
Cancer is a very broad term used to describe a large array of neoplastic diseases. In a 2000 article
proposing six “Hallmarks of Cancer”, Hanahan and Weinberg standardized the steps involved
across cancer types, and described the progression of normal cells to a diseased state following a
succession of hallmark capabilities. Intrinsic to their argument was the idea that all cancer cells
acquire certain traits which cause their transformation and tumorigenesis. The six cancer
hallmarks proposed include: sustaining proliferative signalling, evading growth suppressors,

37
resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating
invasion and metastasis. In theory, these hallmarks all lead to genomic instability, leading a pre-
cancerous cell to become malignant (Hanahan and Weinberg, 2000).
However, more recent work has unravelled additional complexity in cancer development.
In recognition of this, Hanahan and Weinberg published four additional Hallmarks of Cancer
(Figure 4). These include the ability of tumorigenic cells to evade immune destruction and to
reprogram their energy metabolism. However, their report also recognized the increasing
complexity of the “tumor” as opposed to the “cancer cell”, with the tumor having the ability to
change its microenvironment to perpetuate processes such as inflammation that further enhances
genomic instability promoting tumorigenesis (Hanahan and Weinberg, 2011). Nowhere has the
importance of the four additional hallmarks become as important as in the field of cancer
therapy. This is best illustrated in the development of cancer immunotherapies. For instance, the
ability of the tumor microenvironment to upregulate inhibitory molecules, such as CTLA-4 and
PD-1, on cytotoxic T cells has become the focus of several cancer therapies. CTLA-4 is a co-
inhibitory molecule expressed on active CD8+ T cells which generally works to block
proliferation and effector functions of T cells (Walunas et al., 1994). This is achieved by
performing a range of actions, including delocalization of protein kinase C θ and the scaffolding
protein CARMA1 from the immune synapse (Yokosuka et al., 2010), increasing the time of
interaction between the T cell receptor and antigen (Schneider et al., 2005), inhibition of the T
cell stimulatory molecule CD28 by transendocytosis of its ligand B7 (Qureshi et al., 2011) and
enhancement of Treg function (Wing et al., 2008). While expression of the protein is generally
strictly controlled in order to maintain effective immune responses, diseases such as cancer can
result in “chronic” CTLA-4 expression, resulting in a dampened anti-tumour response. This

38
sustained upregulation of CTLA-4 in cancer has become a therapeutic target, with targeted
antibody therapy removing the molecule’s inhibitory effect on T cells and thus allow for the
killing of targeted tumor cells (Grosso and Jure-Kunkel, 2013). Accordingly, CTLA-4 is the
target of two monoclonal antibody-based therapeutics for advanced cancers, especially
melanomas. These drugs are known as tremelimumab and ipilimumab. While tremelimumab
showed regression in 10% of melanoma patients, when delivered with or without a cancer
vaccine (Ribas et al., 2013), ipilimumab showed a regression rate of 21% in melanoma patients
when delivered alone, and a 28% reduction in death risk when delivered with the standard-of-
care drug for melanoma, dacarbazine (Robert et al., 2011). Similarly to CTLA-4, cancer can
result in the chronic upregulation of PD-1, an inhibitory cell surface receptor expressed on CD4+
and CD8+ T cells which generally works to maintain peripheral tolerance following encounter
with its ligand, PD-L1 (Weber, 2010). Similar to anti-CTLA-4 therapy, treatments targeting PD-
1 masks the antigen on the surface of T cells to eliminate its inhibitory effect. The inhibitory
effect of PD-1 has been targeted by monoclonal antibodies, such as nivolumab, which has shown
positive responses in melanoma, non-small cell lung cancer and renal-cell cancer (Topalian et al.,
2012).
While cancer research has previously focused on documenting the genetic changes
occurring within the cancer cell, understanding the host determinants shaping the pathogenesis of
cancer is also important as indicated in the emerging hallmarks by Hanahan and Weinberg. It is
now clear that, Stephen Paget’s reference to the cancer cell “seed” falling on host “soil” has clear
implications in how researchers view cancer, and highlights the need to focus on both cancer cell
and host environment in order to gain a most comprehensive and accurate view of the disease as
a whole.

39
3. CRC Microenvironment
3.1 Intestinal Microenvironment
In the healthy intestine, the base of colonic crypts contain stem cells flanked by niche cells which
regulate stem cell maintenance and normal crypt architecture. In the small intestine, Paneth cells
Figure 4. Hallmarks of Cancer: the Next Generation. With the addition of two additional
Hallmarks, Hanahan and Weinberg acknowledged a role for the microenvironment in the
pathogenesis of most or all cancers. Inflammation by immune cells can lead to the display of
other hallmark characteristics and thus encourage neoplasia. Hypoxic conditions in a tumor
can lead to subpopulations of cancer cells that differ in their method of generating energy,
complementing each others’ metabolic requirements to allow for tumor survival and growth.
(Adapted with permission by Elsevier Ltd: Hanahan D. & Weinberg, R.A., Hallmarks of
Cancer: The Next Generation. Cell 5, 646-674 (2011)).

40
fill the role of the stem cell niche, while an equivalent cell population may exist in the colon
(Sato et al., 2011). Immune cells and vascular endothelial cells also help maintain stem cell
integrity in the crypt by removing aberrant cells and forming extensive vascular networks to
provide nutrients and remove waste, respectively. The regulation of these processes requires the
appropriate secretion of growth factors and chemokines, and is essential for the maintenance of
homeostasis and a normal microenvironment.
During tumourigenesis, mutations accumulate in stem cells, rendering them unresponsive
to suppressive and maturation signals. Conversely, disturbances in the microenvironment can
also trigger cell transformation, leading to uncontrolled proliferation (Figure 5). This can lead to
a positive feedback loop, in which cell-transforming events perturb the microenvironment, which
results in further genetic instability in colon stem cells, eventually leading to colon cancer.
Understanding the changes that occur in the microenvironment during this process could aid in
designing therapeutic interventions which can break this cycle and thus prevent tumor onset.
3.2 Intestinal Stem Cell Niche
In a healthy gut, the stem cell reserve at the base of crypts is necessary to maintain the gut
mucosa, as during maturation, some stem cells move up the crypt as they mature and
differentiate in a continuous cycle of division, differentiation, migration and shedding once the
cells are at the top of the crypts. In the context of cancer however, these cells are susceptible to
transformation, as mutations in colon stem cells can encourage constant cell division and prevent
cell maturation, leading to a tumor cell reservoir . In the colon, niche cells similar to intestinal
Paneth cells act to maintain homeostasis of this cell population, and Paneth cell dysregulation

41
has been associated with inflammatory bowel disease (IBD), which in turn is associated with a
higher risk of developing colon cancer (Clevers and Bevins, 2013). Paneth cells have been
shown in vitro to provide soluble factors such as epithelial growth factor (EGF), transforming
growth factor α (TGFα), WNT3, and the Notch ligand D114. These factors were shown to be
necessary for the expansion of intestinal stem cells, as well as the formation of crypt-like
organoids (Sato et al., 2011). Given that the initiation of sporadic colon cancer often follows
dysergulation of the WNT pathway, it is possible that Paneth cells, or equivalent cells in the
colon, play an integral role in the initial events leading to cancer.
3.3 CRC Stroma
Following the initiation of cancer, the colon microenvironment undergoes drastic changes.
Examination of tissue from breast tumors has described a cancer stroma largely composed of
dense connective tissue, an abundance of fibroblasts, and general remodelling of the extracellular
matrix (Ronnov-Jessen et al., 1996;Tlsty and Hein, 2001). While fibroblasts generally arise from
mesenchymal cells, cancer-associated fibroblasts (CAFs), the term used to specifically describe
fibroblasts found in the tumor microenvironment, can arise from a range of different cell
populations such as mesenchymal stem cells, endothelial cells, adipocytes and cancer endothelial
cells (Zeisberg et al., 2007;Jotzu et al., 2010;Mink et al., 2010). While fibroblasts normally act in
maintaining stromal architecture, through secretion of collagen and extracellular matrix
components, CAFs play a role in multiple steps of cancer pathogenesis, participating in de novo
cancer initiation as well as tumor progression and invasion (Bhowmick et al.,
2004a;Kuperwasser et al., 2004). For instance, the dysregulated release by CAFs of fibroblast
growth factor (FGF), epidermal growth factor (EGF), hepatic growth factor (HGF), macrophage

42
stimulating protein (MSP), colony-stimulating factors (CSF), TGFβ), WNT, matrix
metalloproteinases (MMP), and interleukins can lead to cellular transformation and progression
of a benign lesion into a carcinoma (Bhowmick et al., 2004b) .
Factors such as TGFβ can play complex roles in cancer development and subsequent
metastasis (Simms et al., 2012). The majority of colon cancer cases involve mutations in TGFβ
pathway components, including TGFβ type II receptor mutations (TGFβR2) (Markowitz et al.,
1995;Biswas et al., 2004;Biswas et al., 2008). Interestingly, the effect of TGFβ seems to be
related to its level of expression; at low levels TGFβ is a regulator of stem cell renewal and
differentiation, but at higher levels, it is involved in angiogenesis and immune cell regulation. In
the context of cancer, epithelial cell “miscommunication”, induced by inflammation or tissue
injury, promotes the expansion of stromal fibroblast (Zeisberg et al., 2000), which produce TGFβ
to limit epithelial cell growth (Becker et al., 2004). In 2012, Matise et al. xenografted Tgfbr2
knockout murine mammary carcinoma cells together with mammary fibroblasts on chicken
embryo chorioallantoic membrane in order to model epithelial-stromal crosstalk. They reported
that the Tgfbr2 knockout cancer cells had twice the metastatic potential in this model compared
to WT carcinoma cells. The enhanced metastasis was attributed to increased ability to
extravasate due to downregulation of proteins involved in cell adhesion and tissue maintenance
(Matise et al., 2012). Biswas et al. (2004) observed increased proliferation and neoplasms in
mice with a colon-specific deletion of Tgfbr2 in an azoxymethane-induced colon cancer model.
In an attempt to regain control of excessive epithelial expansion, stromal fibroblasts perpetually
release TGFβ, leading to increased levels which favour tumor growth by encouraging
angiogenesis and immune modulation. TGFβ has also been shown to promote cell proliferation,
survival and metastasis by indirectly upregulating and activating HGF and MSP, which are

43
ligands for the oncogene c-Met, and by driving cancer invasion by indirectly interfering with
Wnt signalling (Vermeulen et al., 2010).
The secretion of immune regulators, specifically cytokines and chemokines, can further
contribute to pathogenesis in the colon. Interleukin-6 (IL-6) is one such cytokine which is
commonly associated with colon cancer, and the levels of which can provide insight into a
patient’s cancer stage and prognosis (Qiao and Wong, 2009;Galizia et al., 2012;Lee et al.,
2013;Lin et al., 2013) . Similarly, the chemotactic protein, Interleukin-8 (IL-8), is found
abundantly in the tumor microenvironment, and has been found to impact tumor initiation and
expansion, metastasis and angiogenesis, largely through a positive JAK/STAT3 pathway
feedback loop (Carpentino et al., 2009).
CAFs are extremely dynamic and well-equipped to alter their behaviour to meet the
needs of tumor cells. During cancer progression, they can mimic myofibroblast activity in tissue
repair, altering the stroma to accommodate tumor expansion (Miles and Sikes, 2014;Narunsky et
al., 2014). Concurrently, their heightened ability to absorb lactate, capacity for lactate oxidation,
and their tendency towards low glucose absorption complements the expression of high levels of
lactate dehydrogenase-5 (LDH-5) and hypoxia-inducible factor 1α (HIF-1 α), high capacity for
glucose absorption and lactate secretion often seen in the anaerobically-inclined cancer cells
(Fiaschi et al., 2012;Guido et al., 2012;Rattigan et al., 2012).
By their ability to promote proliferation, restructure the extracellular matrix and their
ability to accommodate cancer cell expansion, CAFs promote the development of a tumor-
permissive microenvironment in the colon. Understanding the contribution of the stroma to the
promotion of cancer can unveil mechanisms integral in the creation of new therapies targeting
CRC.

44
3.4 Immune Cell Involvement in CRC
The link between cancer and inflammation is now well-established, having first been noted by
Rudlof Virchow in 1863 (Coussens and Werb, 2002), and since having been studied in a diverse
array of carcinomas, including colon cancer. Inflammation is normally a defence response
against invading pathogens and an initiator of wound healing. However, its deregulation is a
usual observation in cancer, with chronic inflammation triggering cancer-causing mutations.
While the immune system can also act to eradicate or control tumorigenesis, certain immune
effectors can inappropriately induce suppression in the tumor microenvironment.
For instance, neutrophils and macrophages can promote processes such as angiogenesis
to promote cancer expansion (Nozawa et al., 2006;Houghton et al., 2010). Macrophages are
especially potent in regulating the inflammatory response, and their polarization in a tumor can
provide prognostic information. For the majority of cancers, an increased presence of
macrophages correlates with more aggressive cancer phenotypes and decreased patient survival
(Nilsson et al., 2012;Zhang et al., 2013a), although this trend may be reversed in colon cancer
(Forssell et al., 2007). This effect is partly the result of a shift in macrophage activity from a
more cytotoxic phenotype towards an immunosuppressive phenotype (Mantovani and Locati,
2013). With this in mind, tumor associated macrophages (TAMs) are divided into groups based
on their polarization. Anti-tumor, or M1 TAMs are cytotoxic, secrete pro-inflammatory
cytokines and express antigen-presenting molecules such as MHC class II, and co-stimulatory
receptors for T cells, making them unfavourable for tumor growth. Conversely, M2 TAMs are
considered to be tumor-promoting, with the capacity to secrete immunosuppressive cytokines,

45
drive cell proliferation, remodel tissues and encourage angiogenesis (Ong et al., 2012;Edin et al.,
2013).
Dendritic cells (DCs) are another immune cell subtype altered in the context of cancer
(Almand et al., 2000). Generally, three subsets of DCs are believed to reside in the tumor
microenvironment. The first subset consists of functionally intact DCs, fully capable of antigen
presentation, the presence of which correlates with improved patient survival . The second subset
consists of DCs with defective antigen uptake, processing and presentation abilities; slower
motility and decreased cytokine release, due to inhibition of DC maturation by factors such as
IL-6 and G-CSF in the cancer microenvironment (Bharadwaj et al., 2007). These cells display an
impaired ability to provide co-stimulatory signals required to initiate effector T cell activation
and proliferation. The last type of DCs seen in the cancer microenvironment have been described
as regulatory DCs with the capacity to suppress T cell proliferation and an ability to polarize T
cells to differentiate to regulatory T cells (Shurin et al., 2013). These cells may impose immune
suppression by secreting a variety of suppressive cytokines and effectors, namely increased
amounts of IL-10, indeoleamine-2,3-dioxygenase (IDO), TGFβ and COX-2 (Ghiringhelli et al.,
2005;Obermajer et al., 2011;Han et al., 2014) and have been associated with reduced patient
survival in colon cancer (Gulubova et al., 2012). Compared to a healthy colon, the colon cancer
microenvironment tends to contain fewer DCs , which display high plasticity and may have
different origins (Shurin et al., 2013).
Another group of myeloid cells, namely immature myeloid precursor cells termed
myeloid derived suppressor cells (MDSCs), have been a subject of intense scrutiny in the field of
cancer microenvironment research. The term MDSC encompasses a wide variety of cell types,
and can be applied to immature neutrophils, monocytes, DCs and early myeloid progenitors.

46
MDSCs have a tendency to drive a suppressive state, seen not only during cancer initiation and
progression, but also in other immune suppressive states, such as that seen in the context of
sepsis. These cells are induced following the release of colony stimulating factor (CSF) by
cancer-associated stroma into the cancer microenvironment, and they effectively inhibit natural
killer (NK) cell activity, cytotoxic CD8 effector T cell expansion and favourably expand the
suppressive regulatory T cell population (Shojaei et al., 2009;Pylayeva-Gupta et al., 2012).
These effects all combine to promote tumorigenesis, and the vascularisation of tumor tissue,
since MDSCs can additionally secrete pro-angiogenic factors such as VEGFA, TGFβ and basic
FGF (Filipazzi et al., 2012). Because the convergence of these effects have such drastic effects
on cancer survival and proliferation, MDSC presence in tumors is generally considered an
indicator of poor prognosis, with a higher percentage of MDSCs present in the tumor milieu
correlating with an increased risk of death in cancer patients (Gabitass et al., 2011).
3.5 CRC Vasculature
As a tumor grows, its capacity to survive and expand depends on its ability to develop its
independent blood supply to provide the cells in the expanding tumor with nutrients and oxygen,
while removing waste. CAFs can, to some extent, complement the metabolic requirements of
cancer cells, by absorbing and oxidizing lactate (Fiaschi et al., 2012;Rattigan et al., 2012).
However, these adaptations remain insufficient, and angiogenesis is still required to meet the
tumor’s metabolic demand. This is supported by research which indicates a direct correlation
between angiogenesis and cancer aggressiveness (Slattery et al., 2014) Because of the essential
role angiogenesis plays in cancer progression (Hanahan and Weinberg, 2000), much research
focused on targeting components of angiogenic processes to treat cancer patients. Unfortunately,

47
the complex nature of angiogenesis in the tumor microenvironment has prevented much progress
in this field. For instance, the use of anti-angiogenic therapies which result in the contraction of
tumor-associated blood vessels can drastically decrease drug efficacy by the very fact that they
destroy the blood vessels used to deliver cancer drugs. As well, these treatments have the
potential to create hypoxic areas inside the tumor that harbour a particular set of chemoresistant
stem cells with low metabolic requirements (Mao et al., 2013). One example of the disappointing
results observed following anti-angiogenic therapy was reported in 2011, after a Phase III trial
with 2672 stage II and III CRC patients showed no improvement following treatment with the
anti-VEGF monoclonal antibody, bevacizumab (Allegra et al., 2011). Previous to this, treatment
of mouse models of pancreatic neuroendocrine carcinoma, glioblastoma, as well as metastatic
models of breast cancer and melanoma with the VEGF/PDGFR kinase inhibitor, sunitinib, or the
VEGFR2-blocking antibody, resulted in increased invasion and metastasis (Ebos et al.,
2009;Paez-Ribes et al., 2009). While the authors believed that this may be due to the selection of
a more aggressive cancer following hypoxia, other mechanisms may account for the resistance to
anti-angiogenic therapy. For example, Cheng et al. revealed that tumorigenic glioma stem cells
injected subcutaneously in mice can give rise to pericytes, cells that wrap around capillaries and
venules and aid in blood vessel formation and remodelling. The authors reported that these cells
aided tumor blood vessel function, tumor expansion and progression (Cheng et al., 2013), and
hypothesized that elimination of these cells could potentially block tumor progression and
improve the efficacy of anti-angiogenic therapy.
Despite the generally disappointing results in cancer treatment with anti-angiogenic
agents, angiogenesis remains a target in several cancer therapeutic strategies. Several of these
treatments, such as bevacizumab, or Avastin, a VEGF inhibitor approved for metastatic colon

48
cancer (Shih and Lindley, 2006), inhibit pro-angiogenic molecules such as EGF, platelet-derived
growth factor (PDGF), transcription factors such as HIF-1α, receptor tyrosine kinases and
MAPK and PI3K signalling components (Shih and Lindley, 2006). Combining anti-angiogenic
drugs with other chemotherapies has improved the success rate of these drugs in CRC, lung,
breast, renal and brain cancers (Mita et al., 2013). In a study with 1401 metastatic CRC patients,
the combination of bevacizumab with oxaliplatin as a first-line therapy improved progression-
free survival by 2.4 months, although survival was not significantly improved (Saltz et al., 2008).
Another class of drugs known as tumor vascular disrupting agents (VDA), uses the disruption of
endothelial cell cytoskeleton or endothelial cell death as a means of destroying tumor-associated
vasculature (Mita et al., 2013). Unlike anti-angiogenic drugs, which are given in the late stages
of cancer, VDAs are provided early on in tumor development, prior to metastasis, in order to
disrupt the newly-forming tumor vasculature and cause tumor necrosis (Gridelli et al., 2009).
This class of drugs is relatively new, but has thus far shown promising results when used in
combination with anti-angiogenic drugs. For instance, the VDA combretastatin A4 phosphate
(CA4P), a microtubule destabilizing drug, in combination with bevacizumab, was shown to
increase progression-free survival by 1.7 months and overall survival by 1.2 months in a cohort
of 15 patients in Phase I clinical trials (Nathan et al., 2012).

49
4. Pre-Metastatic Niche and Organotropism in CRC
Remarkably, the ability of cancer stem cells to favourably alter their environment seems not to
be limited to the immediate environment surrounding a primary tumor, but seems to be extended
to the alteration of microenvironments at distant sites (Sceneay et al., 2013). In situ tumors seem
to be able to distantly manipulate microenvironments, aptly labelled pre-metastatic niches, in
Figure 5. The Primary Tumor Microenvironment. a) Cancer cells in tumors exist in a
complex microenvironment comprising cell types such as stomal fibroblasts, bone marrow-
derived cells, epithelial cells and lymphocytes, which can promote inflammation and
angiogenesis, leading to invasive tumor cells. b) H&E stain of invasive breast cancer tissue
shows infiltration of leukocytes at the tumor site. c) Macrophages in invasive pancreatic
cancer express cathepsin B (green) as cancer cells lose E cadherin (red), allowing for
increased cell motility. (Adapted with permission by Nature Publishing Group: Joyce J.A. &
Pollard, J.W., Microenvironmental regulation of metastasis. Nat. Rev. Cancer 9(4), 239-252
(2009))

50
preparation for metastasizing cells to detach from the primary tumor, travel through the
bloodstream, and set up secondary tumors, or metastases at distant organs (Figure 6). This is seen
in the propensity for certain cancers to favour certain organs for metastases, and in the signalling
between cytokines, chemokines and receptors to predispose tumor cells to home to certain organs
(Joyce and Pollard, 2009). Indeed, Stephen Paget initially noted certain patterns in metastatic
tumor location, the lack of randomness which led to his “seed and soil” hypothesis (Paget, 1889).
For instance, breast cancers have a propensity to preferentially colonize organs such as the lungs,
liver, bone, brain and regional lymph nodes, tissues which all express high levels of stromal cell-
derived factor 1α (SDF-1α), a ligand for the CXCR4 receptor commonly found on breast cancer
cells. While the tumor microenvironment is well understood, the pre-metastatic niche remains
relatively unexplored.
Essential to the creation of a pre-metastatic niche are molecules labelled tumor-derived
secreted factors (TDSFs) and bone marrow-derived cells. Research has shown that different
TDSFs and bone marrow-derived monocytes are required to form the metastatic niches in
different tumor models, with TDSFs such as VEGF and placental-derived growth factor
travelling from the tumor site to mobilize bone marrow-derived cells such as VEGFR positive
hematopoetic progenitor cells in the stroma and extracellular matrix at secondary sites to create
microenvironments that favour their colonization by metastasising tumor cells (Kaplan et al.,
2005). These cells form clusters, expressing the fibronectin receptor integrin VLA-4, which
allows them to interact with local fibroblasts to stimulate fibronectin production and secrete
matrix metalloprotease type 9 (MMP9) to create a favourable environment for disseminating
CXCR4-expressing tumor cells (Kaplan et al., 2005). The creation of pre-metastatic niches has

51
been shown to greatly encourage metastatic growth, though it is still unclear as to whether they
are indispensible in the metastatic process (Sceneay et al., 2013).
While the initial trigger resulting in the release of TDSFs such as VEGF, TNFα, TNFβ,
placental growth factor (PIGF), Lysyl oxidase (LOX), versican and G-CSF is unknown, it is
possible that their secretion arises simply as a result of processes occurring at the primary tumor
site which causes systemic disturbances. For instance, the process of angiogenesis at the initial
site is necessary for the continued growth and survival of a tumor at distant sites (Coussens et al.,
1999;Lin et al., 2006;Nozawa et al., 2006).
Hypoxia occurring at the primary tumor site is another process directly linked to
formation of pre-metastatic niches. As the tumor increases in size, oxygen tissue tension is
reduced to the inadequate blood supply provided by the chaotic blood vessels present in most
tumors. This condition can select for cancer cells which are not only able to survive this
environment, but which have a more aggressive and more invasive phenotype. This may be in
part due to the expression of hypoxia inducible factors (HIFs), some isoforms of which are
associated with increased tumor expansion, angiogenesis and metastasis, as well as poor patient
outcome and propensity to relapse (Bos et al., 2003;Dales et al., 2005). HIFs exert their function
on pre-metastatic niche establishment through LOX and LOX-like families of proteins. These
molecules, which are secreted from hypoxic tumor cells, have been implicated in the remodeling
of the extracellular matrix in pre-metastatic sites, mainly by co-localizing with fibronectin and
cross-linking collagen IV in the basement membrane to promote adhesion of bone marrow-
derived monocytes capable of remodelling the extracellular matrix (Erler et al., 2009). In
addition, hypoxic cancer cells have also been found to be a significant source of TDSFs, such as

52
monocytes chemotactic protein-1 (MCP-1), which promote metastatic niche formation (Sceneay
et al., 2012).
Organotropism associated with the formation and location of the pre-metastatic niche
seems to be dependent on the cancer cell type. This was illustrated by work done by by Hiratsuka
and co-workers in which mice pretreated with media from a B16 melanoma culture prior to
injection of Lewis Lung Carcinoma cells developed metastases in organs predisposed to
metastasis during melanoma, versus metastases preferentially occurring in regions were lung
cancer tends to metastasize (Hiratsuka et al., 2006). Thus, one can conclude that the
organotropism specific to certain cancers is a result of the specific TDSFs secreted by the
primary tumor. MDSCs are frequently found in increased numbers in the pre-metastatic niche
(Hiratsuka et al., 2006;Kim et al., 2009;Kowanetz et al., 2010;Yan et al., 2010;Granot et al.,
2011;Sceneay et al., 2012) and their abundance in cancer is linked to pre-metastatic niche TDSFs
such as S100A8, S100A9, VEGF, MMP9, TGF-β, G-CSF and CCL2. Together these factors
work as chemoattractants to recruit MDSCs to the tumor site (Huang et al., 2007;Shojaei et al.,
2007;Yang et al., 2008) and allow them to undergo restricted differentiation, while limiting their
ability to fully mature.
Similar to MDSCs found in the primary tumor microenvironment, MDSCs in the
metastatic niche display great plasticity, depending on the TDSFs they are exposed to. This is
seen in the differences observed between MDSCs that home to the pre-metastatic niche directly
after leaving the bone marrow, versus MDSCs that reach the pre-metastatic niche after spending
time in the primary tumor microenvironment (Corzo et al., 2010). For example, CD11b+/Ly6G+
myeloid cells that develop into neutrophils at the primary tumor site prevent metastasis, by
eliminating tumor cells in pre-metastatic organs (Granot et al., 2011). Conversely, tumor-

53
secreted factors such as versican can influence CD11b+/Gr-1+ MDSCs in the pre-metastatic
niche to produce TNFα, which enhances tumor cell survival and recruits inflammatory
leukocytes to the pre-metastatic niche (Kim et al., 2009). Like in the tumor microenvironment,
MDSCs in the pre-metastatic niche are linked to immunosuppression due to their ability to
promote Tregs, (Huang et al., 2006;Serafini et al., 2008;Pan et al., 2010) and suppress IFNγ,
resulting in decreased activity of NK, NKT, CD4+ T and CD8
+ T cells (Yan et al., 2010).
Intriguingly, specific subtypes of MDSCs can inhibit pre-metastatic niche formation and gain
anti-tumor activity (Granot et al., 2011).
4.1 CRC Metastasis
Metastasis remains the primary cause of mortality for CRC patients, with 20% of patients
progressing to metastasis, and 40% of patients with localized CRC relapsing with distant lethal
metastases (Tsikitis et al., 2014). In fact, metastasis accounts for 90% of deaths from solid
tumors across cancers (American Cancer Society 2014). Treatment of metastatic-stage cancers
remain tragically inadequate due to incomplete understanding of disease pathogenesis.
Metastasis refers to the growth of secondary malignant tumors at sites distant to the initial
site of cancer. It is an evolutionary process by which cancer cells at the initial tumor can acquire
genetic changes which allow them to survive, proliferate, invade tissues and disseminate to
different organs where they create secondary tumors. While a few cells from the initial neoplasm
detach and invade the stroma, their subsequent entry into the circulation through thin-walled
venules leads to their large-scale destruction, with only a small fraction of survivors. These then
travel to distant organs, where they become entrapped in capillary beds and extravasate to

54
proliferate in the organ parenchyma. Their ability to survive and proliferate in this environment
depends on the capacity to which they can develop a vascular network as well as resist
destruction by host immune and non-immune mechanisms (Chambers et al., 2002)
Figure 6. Tumor Microenvironment Signalling Pathways involved in malignant
progression. Cells in the tumor microenvironment contribute to the tumor and pre-metastatic
niche by maintaining signalling interactions. Interactions between cancer cells, parenchymal
and stromal cells lead to signalling which eventually leads to aggressive cancer phenotypes
such as growth, invasion and metastasis, and may influence distant sites, leading to the
creation of pre-metastatic niches. (Adapted with permission by Elsevier Ltd: Hanahan D. &
Weinberg, R.A., Hallmarks of Cancer: The Next Generation. Cell 5, 646-674 (2011).

55
5. Cancer Immunoediting
The idea of immune surveillance was first suggested 50 years ago, by Burnet and Thomson, who
predicted that the immune system played a role in detecting and eliminating transformed cells
(Burnet, 1957). In the past years, several groups have confirmed this hypothesis, as well as
implicated the immune system in facilitating cell transformation, controlling or promoting tumor
growth. These seemingly paradoxical functions have been integrated into a process known as
immunoediting, a process which encompasses anti-tumor effects, as well as the ability of the
immune system to shape the cancer in three subsequent phases known as Elimination,
Equilibrium and Escape (Shankaran et al., 2001;Dunn et al., 2004;Dunn et al., 2006;Schreiber et
al., 2011).
5.1 Elimination
The elimination phase involves both the innate and adaptive immune systems, which detect and
destroy the initial cancer cells before a visible tumor has formed (Figure 7). Transformed cells
express stress-induced molecules such as calreticulin, NKG2D ligands and tumor antigens
presented by MHC Class I, making them visible to γδ T cells, NK cells and CD8+ effector T
cells. DCs engulf and display tumor antigens, initiating T cell and NKT cell anti-tumor activity
(Bonaccorsi et al., 2014). Subsequent release of IFNγ leads to further anti-tumor effects,
including the suspension of tumor cell proliferation and angiogenesis (Hiura et al., 1994;Luheshi
et al., 2014). The presentation of Fas and TRAIL receptors on tumor cells, can likewise induce
their CD8+ T cell-mediated apoptosis (Grimm et al., 2010). M1-polarized macrophages similarly
keep the tumor under control by secreting IL-1, IL-12, ROS and TNF-α (Costa et al., 2013). The

56
transfer of tumors between WT and immunocompromised mice shows that cancers from mice
lacking recombination-activating gene 2 (Rag2) mice are more immunogenic than tumors arising
in WT animals. Because Rag2 regulates the rearrangement and recombination of
immunoglobulin molecules and the T cell receptor, Rag2-/-
animals which lack functional T, B
and NKT cells, thus providing a tumor microenvironment with less immune selective pressure
than cancers originating from WT mice, with an intact immune system (Kaplan et al.,
1998;Shankaran et al., 2001).These experiments underline the important role that the host plays
in determining the outcome of cancer pathogenesis. While an intact immune system provides
essential protection against cancer, the selective pressure also leads to a less immunogenic
cancer, highlighting the plasticity of cancer cells in adapting to their environment.
5.2 Equilibrium
The equilibrium phase of cancer marks a stage during which the tumor is held in a dormant state
and kept from expanding, although there is no significant decrease in tumor mass either (Figure
8). This is due to the continued genetic and epigenetic changes some tumor cells undergo while
under continuous pressure exerted by the immune system. Thus, some tumor cells mutate to a
state in which they can avoid immune recognition by either decreasing expression of certain
tumor antigens or mutating genes involved in antigen presentation (Udagawa, 2008).

57
Figure 7. Elimination. During the elimination step, the innate and adaptive immune system
work to eradicate clinically undetectable cancers. Stress-induced markers on tumor cells
make them visible to NK and T cells, leading to cytotoxic activity. DCs can also present
tumor antigen, activating further T cell activity. M1-polarized macrophages and granulocytes
can also contribute to tumor destruction by secreting factors such as IL-1, IL-12, ROS and
TNF-α. (Adapted with permission by Elsevier Ltd: Schreiber, R.D. et al., New insights into
cancer immunoediting and its three component phases—elimination, equilibrium and escape.
Curr. Opin. Imm. 27, 16-25 (2014))

58
These cells can also induce immunosuppression by inducing ligands such as PD-L1,
which negatively regulates immune responses when recognized by its cognate receptor on T cells
(Spranger et al., 2013). Thus, the equilibrium stage is essentially a stage at which the anti-tumor
effects of the immune system balance the activity of pro-tumorigenic factors (Vandooren et al.,
2013;Mittal et al., 2014).
For instance, Teng et al. observed that IL-12 and IL-23 showed critical and opposing
roles in maintaining tumor dormancy in mice injected with the chemical carcinogen 3’-
methylcholanthrene (MCA). Other cytokines, such as IL-4, IL-17, TNF and IFNαβ did not play
critical roles during the equilibrium phase, though their importance in the elimination phase of
MCA tumorigenesis is known (Teng et al., 2012).
While NK cells and certain cytokines appear not to play significant roles in maintaining
the functionally dormant state of the tumor, the adaptive immune system and molecules
promoting T helper cell maturation and cytotoxicity such as IFNγ and Il-12 appear to play
prominent roles in maintaining equilibrium. A study by Koebel et al. demonstrated that mice
which developed tumors following injection of MCA were not able to maintain the equilibrium
state following neutralization of CD4, CD8, IFNγ, IL-12, however, animals depleted of TRAIL,
NKG2D or NK1.1 had the same rate of tumour growth and the same time to progression as
control animals (Koebel et al., 2007).

59
Figure 8. Equilibrium. During this stage, the tumor is held in a functionally dormant state,
with balanced activity of anti-tumor and tumor-promoting cytokines. However, cancer cells
undergo epigenetic and genetic changes due to immune pressure, changes which will
ultimately lead to escape. (Adapted with permission by Elsevier Ltd: Schreiber, R.D. et al.,
New insights into cancer immunoediting and its three component phases—elimination,
equilibrium and escape. Curr. Opin. Imm. 27, 16-25 (2014))

60
5.3 Escape
During the escape phase, tumor cells can evade detection and destruction by the immune system
resulting in their ability to expand, invade tissues and metastasize (Figure 9). Mechanisms
leading to tumor cell escape include changes in the tumor cell which lead to reduced immune
recognition, increased tumor cell resistance and survival, or immunosuppression induced by the
tumor microenvironment. Evasion of immune recognition by tumor cells can occur, as
mentioned previously, following mutations which either cause the tumor cell to stop expressing
immunogenic tumor antigen, or cause defects in molecules required for antigen presentation,
such as MHC I, along with the loss of co-stimulatory molecules required to initiate cytotoxic
activity upon antigen recognition (Magner et al., 2000;Liu et al., 2009;Kosmaczewska et al.,
2012). For instance, Ugurel et al. reported that peripheral blood monocytes from 144 melanoma
patients showed decreased expression of MHC class I and class II molecules and the co-
stimulatory molecules CD80/B7-1, which was associated with disease progression (Ugurel et al.,
2004). Cancer cells can also enter the escape phase by increasing the expression of molecules
such as STAT3 or the anti-apoptotic protein Bcl2, which enhance cell resistance and survival. As
well, factors such as IDO, PD-L1, TDO, CD73, galectin-1/3/9, CD39 and adenosine receptors
can combine with the activity of certain MDSCs, M2-polarized macrophages and DCs to induce
immunosuppression, while factors such as VEGF, TGFβ, IL-6, M-CSF promote angiogenesis
(Gajewski et al., 2011;Wu et al., 2013). In general, these factors contribute to the tumor
expansion seen in the escape phase.

61
Figure 9. Escape. In the escape phase of tumor immunoediting, the immune system can no
longer limit cancer cell expansion and the disease becomes clinically significant. Tumor
cells are now able to evade immune recognition, and express molecules which allow for
resistance, survival, immunosuppression and secretion of tumorigenic cytokines. M2-
polarized macrophages, MDSCs and IDO-expressing DCs can stabilize Treg populations
and secrete cytokines which further immunosuppression and allow for tumor expansion.
(Adapted with permission by Elsevier Ltd: Schreiber, R.D. et al., New insights into cancer
immunoediting and its three component phases—elimination, equilibrium and escape. Curr.
Opin. Imm. 27, 16-25 (2014))

62
6. Discovery of Host Genetic Determinants of CRC
6.1 Genetic Screening and Candidate Genes
Several rodent models have been established that reproduce aspects of human CRC. Mimicking
CRC pathogenesis in humans, these models are based on genetic alteration of CRC pathways
such as Wnt/β-catenin, MMR and TGFβ pathways, manipulation of factors such as interleukin-2
(IL-2), IL-10 and T cell receptor α-chain involved in the mucosal immune response, or using the
treatment of carcinogens such as AOM to induce sporadic colon cancer (Boivin et al.,
2003;Rosenberg et al., 2009). However, while these methods can initiate CRC in situ, tumors
from these classical CRC models rarely metastasize, rendering the use of other models necessary
to study metastasis. In 2010, Hung et. al developed a progressive CRC mouse model which
accurately reproduces human CRC progression by generating mice homozygous for a
conditional knockout of Apc and heterozygous for a latent allele of active Kras (Krastm4tyj/+
) (Apc
CKO/LSL-Kras). Administration of adenovirus-cre in these animals resulted in tumorignesis 3
weeks following virus injection, with an average tumor burden of 3.6 tumors per mouse, 64% of
which were adenomas and 36% of which were carcinomas. Following 20 weeks after adeno-cre
injection, 50% of the examined lesions were carcinomas, and at 24 weeks liver metastasis were
observed (Hung et al., 2010). CRC metastasis has also been investigated using alternative
strategies such as intrasplenic, intravenous or orthotopic injections of CRC cell lines to assess
metastatic colonisation of target organs (Zhang et al., 2013b).
While previous work centred on genetic changes occurring within cancer cells, focus on
the role of the host environment in determining the outcome of cancer and metastasis is now
burgeoning. For example, a genetic screen using insertional mutagenesis in Drosophila
melanogaster allowed the identification of genes involved in metastasis. Using tumor phenotype

63
as a readout, the authors were able to identify apontic, involved in formation of the fly eyes and
tracheal system; pointed, required for the formation of glial cells; and semaphorin 5c, involved in
early development, as host genetic determinants of metastasis (Woodhouse et al., 2003;Rollmann
et al., 2007;Zhu et al., 2011;Liu et al., 2014). While mutations in apontic and pointed increased
the severity of metastatic disease, mutations in semaphorin 5c were required for tumorigenesis
(Woodhouse et al., 2003). In mice, Chin et al. used a sleeping beauty transposon in cerebellar
neural progenitor cells to cause systemic dissemination of normally nonmetastatic
medullablastomas in mice heterozygous for Patched, which encodes the inhibitory receptor for
sonic hedgehog (Shh) (Mumert et al., 2012). By determining which genes were located at the
transposon insertion sites, the group was able to identify the genes Eras, Lhx1, Ccrk and Akt as
influencing the metastatic process. Eras is a GTP-binding protein which mimics Ras. Lhx1 is a
transcription factor essential for normal kidney development, and Ccrk is a serine-threonine
kinase which promotes cell cycle progression (Mumert et al., 2012). In previous work, our lab
used a candidate gene approach to show that targeted deletion of genes involved in
inflammasome activation and signalling, namely Nlrp3, Casp1, Il18r and Il18, resulted in a
significant increase in metastatic burden compared to control (unpublished results). Together,
These results highlight the importance of the host microenvironment in modulating the potential
for metastasis and the ability for expansion of aggressive CRC cells.
6.2 ENU Mutagenesis
While reverse genetics involves the study of the effect of a determined genetic change on a
phenotype, forward genetics is phenotype-driven and relies on the mapping of an underlying
mutation. Because this approach mirrors genetic variation in nature, it makes it an unbiased

64
approach for identifying genes involved in the phenotype under study, including poorly-
understood or novel genes.
Germline mutagenesis with the alkylating N-ethyl-N-nitrosourea (ENU) is an efficient
approach to identify host genetic variants which influence disease pathology (Augustin et al.,
2005). A potent mutagen, ENU acts on spermatogonial stem cells to place random single-point
mutations within the mouse genome, making it feasible to identify a single gene responsible for a
particular phenotype. These mutations are mostly A to T transversions and A to G transitions,
with a mutation frequency estimated at about one nucleotide change per million base pairs
(Quwailid et al., 2004), or 1 in 1000 gametes (Rinchik et al., 1990). In total, this represents
approximately 3000 per genome, of which 30 are expected to result in alterations of the amino
acid sequence. Fortunately, ENU mutagenesis typically biases its mutations to occur in coding
regions, splice sites, or in conserved non-coding regions in the proximity of genes (Boles et al.,
2009).
The variants being induced in a known homogeneous background, relationships between
observed heritable phenotypes and the genes causing abnormalities are readily established, even
for genes with previously undefined biochemical and cellular functions. After confirming the
heritability of a deviant phenotype, novel homozygous recessive mutations can be identified via
gene mapping, or more conveniently, cost-effective, and much faster, exome-capture and next-
generation sequencing.

65
Figure 10. A Discovery Platform to Identify Novel Genes that Directly Impact Susceptibility to
Metastasis. ENU-mutagenized males are bred to bring mutations to homozygosity in G3 animals.
CRC metastasis screening identifies deviant pedigrees, and DNA is sent for exome sequencing. Data
analysis and in vivo validation identifies mutations in genes affecting the metastatic process.
Characterization of mechanisms and pathways can lead to novel therapeutic avenues for CRC
treatment.

66
GOALS OF THE STUDY
In this project we used a large-scale ENU forward genetics platform, as well as a
candidate gene approach, to identify host genes which play an important role in the pathogenesis
of CRC metastasis. Injection of C57BL/6 mice with 250,000 colorectal cancer cells typically led
to death 4-6 weeks following injection. We screened for recessive homozygous mutations within
G3 offsprings from ENU-mutagenized mice that lead to susceptibility or resistance to CRC
expansion in the lung, using survival as a readout. In addition, we investigated the role of a
number of immune factors in mediating CRC metastatic growth using mice with targeted gene
deletion (Knockout mice).We aimed to identify novel proteins and biochemical pathways
involved in mediating the CRC metastatic process in order to better understand the disease and in
order to identify possible therapeutic targets for treating metastasis (Figure 10).

67
MATERIALS AND METHODS
Mice
All mice were maintained at McGill University and bred on a C57BL/6 background. ENU mice
were generated in the facility and animals were used when at 6-8 weeks old. WT animals and
Rag1-/-
, Casp1-/-
, Casp12-/-
, Ripk2-/-
, Ripk3-/-
, Birc3-/-
, Bid-/-
mice were described previously
(Mombaerts et al., 1992;Kuida et al., 1995;Chamaillard et al., 2003;Newton et al., 2004;Conte et
al., 2006;Ness et al., 2006;Saleh et al., 2006)). These mice were used for the CRC candidate gene
screening approach. They were also injected with MC38met-Luc at 6-8 weeks of age. Jak3W81R
mice for both screening approaches were kindly provided by Dr. Phillippe Gros (Bongfen et al.,
2012). All animal experiments were approved by the McGill University Animal Care and Ethics
Committee in accordance with the guidance of the Canadian Council on Animal Care.
Generation of ENU Mutants
ENU-mutagenized male mice were out-crossed with WT C57Bl/6 females from our facility to
produce G1 offspring which contain one set of mutagenized chromosomes, as well as one set of
wild-type chromosomes. Individual G1 males were then used to establish individual pedigrees to
bring ENU-induced sequence variants to homozygosity. This is achieved by breeding G1 males
with wild-type females to generate G2 mice, and subsequently mating G2 females with their G1
father to produce G3 offsprings. With this scheme, approximately 50% of ENU-induced variants
of G1 males are inherited by each G2 daughter, and 25% of these are expected to come to
homozygosity in G3 offsprings. About 12.5% of G3 offspring are expected to be homozygous
for an estimated 4 functional recessive mutations.

68
Model of CRC Metastasis
Mice were injected with 250,000 highly metastatic MC38 mouse colon adenocarcinoma cells,
which are syngeneic with C57BL/6 mice. The cells were obtained from Vanderbilt University,
and had been selected for their metastatic potential and engineered to stably express the firefly
luciferase reporter gene (MC38met-Luc) (Smith et al., 2010). Injection of the cells into the tail
vein resulted in colonization of the lung and subsequent tumor growth. Following injection of the
MC38 cells, bioluminescent imaging was used to ensure the cells reached the lung. Animals
were then housed in the Goodman Cancer Centre animal facility until endpoint, and survival was
recorded.
MC38met-Luc Cell Culture
MC38met-Luc cells were cultured for 2 weeks prior to injection, in 20 ml of RPMI media
(Wisent Inc.) supplemented with 10% Fetal bovine serum (FBS; Wisent Inc.), 1% L-glutamine
(Gibco; 200mM), 1% Penicillin-Streptomycin (Gibco; 10,000 U/ml; 10,000 µg/ml) and 40 µl of
G418 (Sigma; 50 mg/ml) to maintain luciferase expression. Cells were initially thawed in 12 ml
of supplemented media in a 10 mm plate, then transferred to a 20 mm plate three days after
thawing. The media was changed 24 h after thawing cells. For passage, cells were incubated for
3-5 minutes at 37°C in 3 ml of trypsin (Gibco; 0.25%). Cells were split every 3 days until
injections. Prior to injection, cells were counted and resuspended in PBS (Wisent Inc.) at a
concentration of 1.25 x 106 cells/ml. Each mouse was injected with 0.2 ml of this solution, to
receive 2.5 x 105 cells each.

69
ENU Screen
Approximately 60-100 G3 animals were injected with MC38met-Luc cells each week, as G3
offspring breeding schedules allowed. In total, at least 16 mice were screened per pedigree, in at
least three different injection rounds. Interestingly, in a previously performed experiment, our lab
had found that ENU-generated mice with a loss of function point mutation in Jak3 kinase
(Jak3W81R
mice), which have reduced numbers of T, B and NK cells, were highly susceptible to
MC38 lung tumor growth (Bongfen et al., 2012). Thus, each injection round included WT
controls and Jak3W81R
mice as a positive control for susceptibility. ENU G3 mice which died
earlier than WT mice were considered susceptible, while those dying after WT mice were
considered resistant.
We screened 500 G3 offsprings from 49 female G2 mice derived from 39 G1 pedigrees over the
course of 12 months. Families containing mice which were considered potentially “susceptible”
or “resistant” were screened in multiple injection rounds to validate the phenotype and to
confirm heritability of the trait in question.
DNA Extraction and Purification
The tail of each G3 mouse was kept in a -80°C freezer. Following confirmation of families
displaying an interesting, heritable trait, tails were digested with proteinase k (Thermo Scientific,
20 mg/ml) in tail lysis buffer (1 M Tris pH 8, 3 M KCl, 0.5 M EDTA, Igepal, Tween20, ddH20)
overnight, before DNA was extracted. DNA was extracted by adding 500 µl of
phenol:chloroform (Fisher Scientific):isoamyl alcohol (Bioshop) (PCI) (25:24:1) to each sample,

70
followed by 20 minutes of shaking at room temperature, and subsequent centrifugation for 10
minutes at 13,000 rpm at room temperature. The aqueous phase was then transferred to a new
tube, where the process was repeated, and then repeated once more using chloroform:isoamyl
alcohol 24:1 mixture instead of PCI. DNA was then precipitated from the aqueous phase with 1
ml of isopropanol and then re-suspended in 200 µl of TE buffer (10mM Tris-Cl pH 7.5, 1 mM
EDTA) for 30 minutes in a 65°C incubator. DNA was stored at 4°C until sent for exome
sequencing. DNA quality was assessed by running DNA samples on a 0.7% agarose gel
(Bioshop) and staining with 2 µl EtBr (Bio Basic Inc; 10mg/ml) .
Exome Sequencing
DNA samples from three affected mice/family for 2 families displaying resistance to MC38met-
luc proliferation were sent to the Australian Phenomics Facility (APF) for exome sequencing.
Extracted DNA was sent from McGill to APF, where the samples underwent exome enrichment
(Agilent SureSelect XT2 All Exon Kit), followed by sequencing by an Illumina HiSeq 2500 as
Paired Ends 75 base pair reads. Reads were then aligned to the APF ENU reference genome with
Burrow-Wheeler Aligner (BWA) software. Raw SNPs were then compared to the reference
genome using Sequence Alignment/MapTools (SAMtools) to exclude known variation in the
samples (eg. dbSNPs, common exome variants, etc.). The remaining SNPs were then filtered for
coding or splicing variants by aligning sample exomes to Ensembl and were filtered for non-
synonymous variants using Anotate Variation software (ANNOVAR). Genes containing multiple
single nucleotide variants (SNVs) were then removed from the list of candidate SNVs.

71
Antibody Depletion
NK cell depletion was achieved by intraperitoneal (i.p.) injection of rabbit anti-mouse/rat Asialo
GM-1 antibody from Cedarlane (clone 8955) that was administered at 250 µg every 5 days until
endpoint, beginning the day prior to MC38 injection. ILC depletion was achieved by i.p.
injection of anti-CD90.2 mAb (30H12) from BioXCell that was administered i.p. at 200 µg every
3 days, beginning one day prior to MC38 injection.
Lung Digestion/Cell Isolation for Flow Cytometry
Lungs were dissected from animals and immediately placed in 1 ml of Medium B (RPMI; 10mM
HEPES; 2mM EDTA; 2-β-mercaptoethanol, 1ul/ml; 1 M Ca2+
). Lungs were then chopped into
small pieces and incubated at 37°C for 35 minutes in 5 ml of digestion media (Collagenase II,
100 mg/ml; DNAse I, 40 U/ml; 1 M Ca2+
) on a bactoshaker. 10 ml of Medium B was then added
to each tube to stop the collagenase reaction, and lungs were gently passed through a 16G needle
10 times, prior to being filtered into a 50 ml tube through a 70 micron strainer. The samples were
then centrifuged at 1500 rpm for 5 minutes, before being suspended in 3 ml of ACK buffer (1.5
M NH4Cl, 100 mM KHCO3, 10 mM EDTA-2Na) for 2 minutes, before being diluted with
Medium B reaching the top of the tube and spun at 1500 rpm for 5 minutes. The supernatant was
then aspirated and resuspended in 1 ml cold PBS, with 2% sterile FBS.
Flow Cytometry

72
Collagenase-digested lungs were surface stained with a combination of monoclonal fluorescently
conjugated antibodies: Vivid V500 (molecular probes), CD3e PerCP-Cy5.5 (eBioscience), CD25
PE (BD), NK1.1 APC (BD), CD11c PE-Cy7 (BD), TCRb PerCP-Cy5.5 (eBioscience), CD11b
eFluor450 (eBioscience), CD90.2 APC-Cy7 (BD), CD127 AlexaFluor 488 (BD), B220 V500
(BD). Antibodies were all kept at a concentration of 0.2 µg/µl, and diluted in PBS supplemented
with 4% FBS. The amount of each antibody per 1x106 cells is: Vivid (0.25µl), CD3e (0.13 µl),
CD25 (0.2 µl), NK1.1 (0.13 µl), CD11c (0.06 µl), TCRb (0.13 µl), CD11b (0.06 µl), CD90.2
(0.03 µl), CD127 (0.6 µl) and B220 (0.25 µl). Samples were fixed in PBS containing 1%
formaldehyde and data acquired on a Canto instrument (BD Biosciences). Data was analyzed
using FlowJo software.
Bioluminescence Imaging
Mice were imaged every 5 days, beginning on the day of injection. Mice were anaesthetized
using 2% aerosolized isofluorane (Baxter Corp.) and subsequent imaging was done following the
intraperitoneal injection of 50 µl of luciferin (30 mg/ml) using a Xenogen IVIS 100 system,
taking images every minute until the peak activity of the luciferase was reached and the peak
intensity was recorded.
Histopathology
Lung lobes were collected in 10% neutral-buffered formalin and tissue sections were prepared
from paraffin block and stained with Hemotoxylin and Eosin (H&E). Images were scanned with

73
a Zeiss LSM Pascal on Axiovert 200 microscope and metastases were counted using Spectrum
(Aperio Technologies Inc.), a database system used to house, manage and analyze whole slides.

74
RESULTS
1. Candidate Genes
Our lab has previously demonstrated that mice with targeted deletion of specific innate immunity
effector genes such as Casp1, Il18 or Il18r1 were susceptible to MC38 colonization in the liver
when injected intrasplenically (unpublished data). We sought to determine the effect of several
immune pathways in regulating the survival following MC38inv tail vein injection. Tail vein
injection was used to model CRC lung metastasis, which affects up to 20% of CRC patients
(Villeneuve and Sundaresan, 2009), since MC38inv-Luc cells pass directly from the blood
stream to the lung and can thus easily access and colonize this organ. Interestingly, Casp1
deletion did not recapitulate the data seen in our lab’s liver metastasis model, with approximately
85% of these mice reaching endpoint during the same time as WT. Similarly, mice lacking
Casp12, a caspase-1 related protein involved in ER stress-induced cell death (Nakagawa et al.,
2000) and a negative regulator of caspase-1 (Saleh et al., 2006) and NF-B activity (LeBlanc et
al., 2008;Labbe et al., 2010) did not exhibit differential survival compared to WT mice (Figure
11A), with all animals succumbing to MC38met-Luc expansion by day 53.
We also examined the effect of NOD1/2 signalling pathway components Birc3 (cIAP2)
and Ripk2 (Rip2) on survival following MC38 lung colonization (Figure 11B). Once again, no
effect was seen following MC38 i.v. injection in mice lacking functional copies of these proteins,
with Ripk2-/-
mice succumbing to metastasis at the same time as WT by day 28 post-injection,
and 80% of Birc3-/-
mice succumbing at the same time as WT around day 43. Two of the Birc3-/-
animals screened survived for 60 days, however this was likely due to variation in the MC38met-
luc cells themselves, or possibly variation in injection technique. Similarly, no difference was

75
observed for mice lacking the cell death effectors Rip3, with Ripk3-/-
mice dying along with WT
between days 28 and 35, and Bid-/-
mice succumbing alongside WT animals from days 30 to 58
(Figure 11C). However, mice with an inactivating point mutation in the Jak3 gene, which
encodes Janus kinase 3 (Jak3), an enzyme involved in signal transduction predominantly
expressed in hematopoetic cells, were found to be significantly susceptible to MC38inv injection
compared to WT mice. Jak3W81R
mice are severely depleted of CD8+ T, B cells and NK cells,
and have a non-functional CD4+ T cell compartment (Bongfen et al., 2012). Rag1-/-
mice were
found to be susceptible to MC38 lung colonization compared to WT control, surviving for only
25 days as opposed to the 30 days that WT survived, they survived significantly longer than
Jak3W81R
mice, which all succumbed by day 20, indicating that protective factors might be
present in these mice which Jak3W81R
mice lack (Figure 12A). Lung weight and histological
analysis on day 15 post MC38 injection also revealed lower lung weight average of 0.5 g for
Rag1-/-
mice compared to an average lung weight of 0.7 g in Jak3W81R
mice (Figure 12B), and a
trend towards lower lung metastasis coverage of approximately 60% coverage, versus the
metastasis coverage two of three experimental Jak3W81R
animals displayed (Figure 12C),
although in both assays Rag1-/-
mice were more susceptible compared to WT mice. Measurement
of luciferase activity every five days post-injection showed an increased photon count in
Jak3W81R
mice beginning from day 0, and continuing until endpoint, again underlining
susceptibility compared to Rag1-/-
and WT mice (Figure 12D). We concluded that Rag1-/-
mice,
which lack a functional adaptive immune system, but still maintain intact innate immune
responses, are resistant to MC38 lung colonization compared to the more severely
immunocompromised Jak3W81R
mice, due to potential NK cell or innate lymphocyte (ILC) anti-
tumor functions.

76
Recent work has highlighted the contribution of innate lymphoid cells (ILCs) in Rag1-/-
mice in the context of diseases ranging from influenza infection (Monticelli et al., 2011) to
colorectal cancer progression (Kirchberger et al., 2013). Using flow cytometry, we ascertained
that that Rag1-/-
lungs contain approximately 2 x 104 functional NK cells and 1 x 10
3ILCs, while
Jak3W81R
mice contain negligible numbers of both cell types (Figure 13 A, B). Thus, we sought
to investigate the relative roles of these cell compartments in the context of MC38inv cell
colonization in the lung.
In order to achieve this, we used monoclonal antibodies to deplete different cell types;
anti-Asialo GM1 to deplete NK cells and anti-CD90.2 to deplete ILCs from Rag1-/-
mice (Figure
13 B-E) .Given that anti-CD90.2 antibody depletion was expected to partially deplete NK cells
as well, depletion of NK cells alone was completed to control for this. Depleting antibodies were
injected every 3 days, beginning 3 days prior to MC38met-Luc injection. NK cell-depleted mice
were sensitized to MC38 lung colonization, and reached endpoint significantly earlier than PBS-
treated controls, dying by day 18, compared to the 23 days that PBS-treated Rag1-/-
animals
reached. (Figure 14A). This heightened susceptibility to MC38 expansion was re-capitulated in
data acquired on day 15 post-injection, with NK cell depleted mice showing a significantly
higher tumor burden compared to WT and ILC-depleted mice (Figure 14B, C), with a
significantly different average lung weight of 0.9 g compared to the 0.5 g average lung weight of
PBS-treated Rag1-/-
mice (Figure 14B) and an average metastatic coverage of 70% compared to
the 35% metastatic coverage of PBS-treated Rag1-/-
mice. Furthermore, depletion of NK cells
resulted in Rag1-/-
mice phenocopying Jak3W81R
mice, indicating that the absence of NK cells in
Jak3W81R
mice may account for their extreme sensitivity and susceptibility to MC38 expansion.
While the lung weight of Jak3W81R
mice was lower than that of NK cell-depleted Rag1-/-
mice, at

77
an average of 0.6 g, the metastatic coverage was heightened, at around 60% coverage, compared
to the average 40% coverage observed in PBS-treated Rag1-/-
animals (Figure 14C), and
Jak3W81R
animals survived until day 18, the same time point that NK cell-depleted ILCs do not
seem to play a role in explaining this phenomenon, having no effect on Rag1-/-
mouse survival,
with CD90.2-treated Rag1-/-
dying between days 20-23, similar to PBS-treated Rag1-/-
. Lung
tumor burden (Figure 14B,C) following MC38inv injection was also not significantly increased
in CD90.2-treated animals compared to CT, with an average lung weight of 0.6g and metastatic
coverage of 50% on Day 15 post-injection .
2. ENU Screen for Host Genetic Determinants of CRC Metastasis
In addition to our candidate gene approach, we used a forward genetics approach towards
identifying host genetic determinants of CRC metastasis. This approach mirrors the genetic
variation seen in nature and thus allows for the unbiased discovery of potentially novel genes
which can affect CRC metastasis.
We decided to use the ENU mutagenesis approach, in which the alkylating N-ethyl-N-
nitrosourea (ENU) induces random single-point germline mutations within the mouse genome.
This characteristic of ENU makes it feasible to identify a single gene responsible for a particular
phenotype and an efficient method for identifying host genetic variants which influence disease
pathology (Augustin et al., 2005).These mutations are mostly A to T transversions and A to G
transitions, with a mutation frequency estimated at about one nucleotide change per million base
pairs (Quwailid et al., 2004), or 1 in 1000 gametes (Rinchik et al., 1990). In total, this represents
approximately 3000 per genome, of which 30 are expected to result in alterations of the amino
acid sequence. Fortunately, ENU mutagenesis typically biases its mutations to occur in coding

78
regions, splice sites, or in conserved non-coding regions in the proximity of genes (Boles et al.,
2009).
ENU-mutagenized male mice were out-crossed with WT C57Bl/6 females from our
facility to produce G1 offspring which contain one set of mutagenized chromosomes, as well as
one set of wild-type chromosomes. Individual G1 males were then used to establish individual
pedigrees to bring ENU-induced sequence variants to homozygosity. This is achieved by
breeding G1 males with wild-type females to generate G2 mice, and subsequently mating G2
females with their G1 father to produce G3 offsprings. With this scheme, approximately 50% of
ENU-induced variants of G1 males are inherited by each G2 daughter, and 25% of these are
expected to come to homozygosity in G3 offsprings. G3 offspring were tested for resistance or
susceptibility to MC38met-Luc injection over the course of at least 4 injection rounds, with a
total of at least 16 animals tested per family.
Screening of 39 ENU pedigrees resulted in the discovery of two families, pedigrees 13
and 31, which consistently displayed metastasis-resistant mutants in a 1/4 Mendelian ratio
(Figure 15). Overall, 500 G3 mutant mice were screened, from 39 G1 males, with the discovery
of 2 deviant pedigrees (Figure 15A). Each family was tested for a minimum of four rounds, with
at least 16 animals tested in total to confirm heritability of the phenotype, with these rounds then
compared in order to determine the presence of a genetically-based deviant phenotype. The ENU
screen was based on survival following tail-vain injection of 250,000 MC38met-Luc cells in
order to model CRC lung metastasis, and animals were screened for resistance or susceptibility.
Mice surviving longer than WT animals were considered resistant and those succumbing in the
same time frame as Jak3W81R
mice were considered susceptible. Several pedigrees exhibited
“deviant” phenotypes in one or two injection rounds, but did not recapitulate this phenotype in

79
subsequent rounds (Figure 15B) and were thus not considered deviant. As well, in order to
identify a genetic basis for a deviant phenotype, families with “deviant” animals were examined
to see if the deviant phenotype segregated into the 1/4 ratio expected when screening for a
homozygous recessive mutation. The two families which were identified as potential deviants all
displayed an approximately 25% penetrant resistant phenotype in at least 3 injection rounds.
Family 13 did not display a phenotype in injection round 1 (Figure 15C), but 22% of all animals
screened in the subsequent injection rounds were resistant compared to WT mice, with one
animal in injection round 3 surviving until day 70 and surviving two weeks longer than the WT
control (Figure 15C). Family 31 was screened in 6 injection rounds, and did not display
resistance until round 3 (Figure 15D). Overall, 20% of animals showed a resistant phenotype,
suggesting the possibility of a homozygous recessive gene influencing this phenotype.
Following exome sequencing, and cross-comparison of samples, we have assembled a list
of potential gene mutations that might underlie the observed deviation in survival for each
pedigree. For pedigree 13, this list includes 4930430F08Rik and Ap1g2 (Table 2).
The candidate, 4930430F08Rik, encodes a protein on chromosome 10, which is currently
uncharacterized, but was first identified by a genome-wide transcriptome analysis by in situ
hybridization of the developing mouse embryo at day 14.5 (Diez-Roux et al., 2011). Our analysis
revealed a novel guanine to adenine point mutation in exon 5 resulting in a serine to leucine
switch at position 187 (Figure 16). This could have implications for the protein encoded by this
gene, possibly causing a disruption in protein three dimensional conformation due to the switch
from the nucleophilic serine to the hydrophobic leucine, or potentially due to the removal of a
key phosphorylation site. This mutation appeared in all three deviant mice samples sent for

80
exome sequencing. It was found in homozygosity in two affected mice however the third
affected mouse was heterozygous for this mutation.
The second candidate for family 13 appeared as a cytosine to adenine point mutation in
exon 14 of Ap1g2 on chromosome 14, changing the arginine at position 459 to a leucine (Figure
17), and was shared between two deviant members of the family, although it appeared as a
homozygous mutation in one animal and was heterozygous in the other (Table 1). The third
mouse was a WT. Adaptor-related protein complex 1 gamma 2 Subunit (Ap1g2) encodes a
gamma adaptin protein. Adaptins are proteins that interact with membrane-bound receptors to
help form the clathrin-coated vesicles which transport proteins from the membrane or the trans-
golgi network to the lysosomes (Boehm and Bonifacino, 2001). Together with other proteins,
adaptins can form a heterotrimeric complex known as an adaptor, to allow the formation of
clathrin-coated vesicles. Ap1g2 is believed to function at a trafficking step in the protein
shuttling pathway between the trans-golgi network and the cell membrane (Takatsu et al., 1998).
While relatively uncharacterized, and unstudied in the context of cancer, the Immunological
Genome Project database lists Ap1g2 as being expressed highly in CD8 effector and memory T
cells, as well as in B cells in the peritoneal cavity. The protein has also been shown to interact
with the L protein of hepatitis B virus, indicating a role in viral pathogenesis (Hartmann-Stuhler
and Prange, 2001). Another group has demonstrated the ability of Ap1g2 to interact with the E3
ubiquitin ligase Neural precursor cell expressed developmentally down-regulated protein 4
(Nedd4) via a ubiquitin-interacting motif, possibly indicating a role for Ap1g2 in the
multivesicular body pathway that is different from that of other adaptins (Rost et al., 2008). The
point mutation found in our deviant animals changes the large and basic arginine residue to a

81
small and hydrophobic leucine residue, thus could have detrimental effects on protein
conformation, leading to changes in protein activity.
Analysis of Pedigree 31 identified four mutations common to all deviants analyzed.
These included mutations in Nbeal1, A130010J15Rik, Cadm3 and Ripk2 (Table 2). Interestingly,
three of the above-mentioned mutations were found on chromosome 1, a promising indicator that
the mutation is in fact part of an island of genes from the initial ENU-mutagenized male. Thus,
the mutation causing the deviant phenotype in pedigree 31 likely resides on chromosome 1,
although it remains to be seen which specific gene is mutated.
The first candidate, neurobeachin-like 1 (Nbeal1), displayed an adenine to guanine point
mutation in exon 35 (Figure 18) which appeared in all three deviant samples, although it was
found to be homozygous in only one sample, and heterozygous in two animals (Table 3). This
mutation led to a serine to glycine amino acid substitution which was deemed to be probably
damaging by PolyPhen SNP data collection predictor, possibly due to the change in amino acid
size or polarity caused by this substitution. First identified in 2002, the gene remains poorly
understood, but was named for its similarity to neurobeachin (Nbea), a protein which is believed
to play a role in protein trafficking in neuronal cells and is able to bind the regulatory subunit of
protein kinase A (PKA) (Wang et al., 2000). Nbeal1, which also contains a Beige and Chediak-
Higashi syndrome (BEACH) domain, is believed to play a role in protein-binding, and has been
found to be expressed in various tissues and cells such as in neurons in the brain and spinal
chord, as well as in macrophages, DCs and B cells (GeneCards, 2014). The Human Malady
Compendium has associated the gene with Lateral Sclerosis (Hadano et al., 2001), and
interestingly, ovary serous adenocarcinoma, and links have also been drawn to lung cancer by
the Asian Bioinformatics Research and Education Network (Gene Expression Omnibus, Dataset

82
GSE31210, 2011). The protein has been found to be up-regulated in glioma and various other
cancers (Table 4) (Chen et al., 2004).
Also on chromosome 1, a thymine to cytosine point mutation in exon 2 of
A130010J15Rik leading to a substitution of a valine to an alanine was found in all three samples
(Figure 19, Table 2). This mutation, which was found to be homozygous in two of the sequenced
samples and heterozygous in the third animal, is possibly damaging to the protein structure,
possibly due to the insertion of a hydrophobic valine in place of the small glycine. The gene is
not well characterized, but has been shown to be highly expressed in epidermal DCs according to
the immunological genome project (Heng and Painter, 2008).
The last gene on chromosome 1 to contain a potential causative mutation is cellular
adhesion molecule 3 (Cadm3). Found in all three samples, the mutation was homozygous in two
mice and heterozygous in the third. It results in a cytosine to adenine switch in exon 8 and leads
to an alanine to serine substitution (Figure 20, Table 2), was deemed to be possibly damaging by
PolyPhen prediction. Cadm3 has been identified as a tumor suppressor in human non-small cell
lung cancers and encodes a transmembrane glycoprotein with an extracellular domain
homologous to that of immunoglobulin superfamily proteins (Fukuhara et al., 2001). Cadm3 has
also been called nectin-like 1 (Necl-1) for the similarity it displays to nectins, proteins which are
Ca2+
–independent immunoglobulin proteins that play a role in the organization of epithelial and
endothelial junctions via three immunolglobulin-like domains in the extracellular compartment
involved in cell-cell adhesion. Thus, Cadm3, which has also been shown to be specifically
expressed in sensory and motor neurons, similarly has Ca2+
-independent hemophilic and
heterophilic adhesion activity. Under normal conditions, it has been shown to participate in the
formation of synapses, axon bundles and myelinated axons (Dong et al., 2006). In addition to

83
being identified as a tumor suppressor in lung cancer (Fukuhara et al., 2001), Cadm3 expression
has been reported to be lost in gliomas (Gao et al., 2009) and mice lacking Cadm3 exhibit
delayed myelination (Table 4) (Tanabe et al., 2013). While the protein has mainly been studied
in neural tissue, it is also highly expressed in spleen CD4+ DCs, and to a lesser extent in
monocytes and epidermal DCs (Table 4) (Heng and Painter, 2008).
Exome sequencing data also revealed a common mutation on chromosome 4, in the gene
encoding Receptor-interacting serine/threonine kinase (Ripk2) (Figure 21, Table 3). This
receptor-interacting protein contains a caspase-recruitment domain which allows it to participate
in the NOD1/2 signalling pathway described above. The mutation was found to be heterozygous
in two mutants and homozygous in one. It causes an adenine to guanine base change which leads
to a disruption in splicing. Ripk2 has been implicated in a number of diseases, and has already
been linked to several cancers, including a role for promoting breast cancer metastasis (Singel et
al., 2014) (Table 4) and has been shown to protect against inflammation-induced colon cancer in
mice (2013;Couturier-Maillard et al., 2013). Given that injection of Ripk2-/-
mice with MC38met-
Luc did not lead to a significant difference in survival compared to WT mice (Fig. 11), it is
possible that this candidate mutation, if causative, does not mimic the effect of a knockout.
Alternatively, the proposed ENU mutation in Ripk2 might lead to deregulation of protein levels,
or could alter the CARD modifying affinity with interacting proteins. Further investigation is
warranted to determine the exact effects of this mutation on the activity of Ripk2.
Following the identification of short-listed genes for both families 13 and 31, we
attempted to confirm which causative gene underlies the observed phenotypes. Genotyping
primers were designed for the short-listed genes, and both deviant and non-deviant mice were
sequenced to determine whether a candidate mutation segregated within the deviants of the

84
family, without being found in the non-deviant members. These experiments are currently
ongoing.
DISCUSSION
Our lab has previously shown the importance of the host environment in the regulation of CRC
metastasis, and the above-mentioned work again emphasizes the impact that host genetics can
have on disease outcome. While the candidate gene approach has many merits, and has allowed
us to study the importance of different immune cell contributions in the context of CRC
metastasis to the lung, the addition of a forward-genetics approach provides us with the potential
to unveil functions in CRC metastasis in genes that may be unknown, or as of yet unstudied.
Because our lab had previously found a role for innate immunity factors such as Casp1 in
regulating CRC progression (unpublished results), we decided to test KO animals for Casp1,
Casp12, Ripk2, Birc3, Ripk3 and Bid in our metastatic lung model. None of the KO animals
showed any significant difference in survival when compared to WT mice. This could be due to
the potency of the MC38met-Luc in our model, which may expand too aggressively or quickly
for these innate immunity factors to control. In fact, the intrasplenic injections in our lab which
found a role for Casp1 in controlling MC38 injection was done with MC38-Luc cells, rather
than the highly invasive MC38met-Luc cells that were used in our CRC lung metastasis model. It
is possible that in a less aggressive model, Casp1 may play a role in controlling MC38 expansion
in the lung as well as in the liver. As well, differences between the microenvironment in the lung
and the liver may account for the observed differences in MC38 expansion between the two
models. Lastly, it is possible that innate immunity factors play a role in preventing cancer

85
development prior to the stage of disease that our model imitates. For instance, in a mouse model
of bladder cancer metastasis, Ripk2-/-
mice were observed to develop larger tumour than WT, and
Ripk2-deficient tumours were found to have enhanced epithelial-to-mesenchymal transition and
an increased number of MDSCs. Thus, there are many stages of cancer which our model does
not encompass, and the fact that the candidate genes we screened did not affect survival in our
model does not preclude that they may play a role in different stages of the metastatic process, or
play a role in cancer control in different organs or in the control of other types of cancer.
While the CRC lung metastasis work described in this thesis is representative of an
aggressive metastatic model, it is important to remember the progressive nature of cancer, and
the length of time over which the initial tumor expands, all the while creating its unique niche
within the host. Sporadic cancers can take over 20 years to develop, with many changes
occurring along the way that influence the transformation of cells and the microenvironment
these cells find themselves in. Our model is unable to replicate this slow process of cell
transformation and expansion to clinical significance, and may therefore miss key players which
are involved in cancer pathogenesis at these early stages. While metastases become established
much faster than the primary tumor from which they originate, the process is also gradual in
comparison to our model, and priming of the metastatic niche may occur before the cells even
reach the site of metastases. Thus, the model used in this work may be useful in determining host
factors important for control of metastases after colonization, but may not provide insights into
factors aiding in the establishment of the pre-metastatic niche and metastasis organotropism. As
well, the model described in this work is representative of aggressive CRC metastatic cancer
behaviour, but may be too aggressive to allow for the detection of host factors with smaller
contributions to cancer. Thus, although this work has not found a role for ILCs in the context of

86
our model, a more gradual, less aggressive model may provide a more accurate idea of whether
these cells play a role in the control of cancer and metastasis. For instance, the progressive model
proposed by Hung et al. would be a better option for host factors of CRC metastasis, as it
faithfully reproduces the steps involved in human CRC progression, including the eventual
metastasis that follows tumor development and growth (Hung et al., 2010). These Apc CKO
LSL-Kras CRC mouse model is excellent because it recapitulates the genotype-phenotype CRC
progression model which occurs in humans. As in human CRC, these metastases primarily
colonize the liver (Derry et al., 2014), however, even this model does not reproduce the lung
metastases found in many CRC patients, a caveat for those studying the lung as a metastatic site.
As of yet, no in situ model of CRC metastasis to the lung exists.
Using the candidate gene approach, our lab was able to shed further light on some of the
important players in CRC pathogenesis. Tail vein injection of MC38met-Luc cells into Rag1-/-
mice led to an intermediate survival compared to susceptible Jak3W81R
mice, leading us to
investigate the innate factors in Rag1-/-
mice which led to their relative resistance. Realizing that
Rag1-/-
animals still contained ILCs and NK cells, unlike Jak3W81R
mice (Figure 13A), we sought
to deplete both populations in Rag1-/-
animals injected with MC38met-Luc and discovered that
NK cells primarily account for the control of MC38met-Luc expansion in Rag1-/-
animals, with
ILCs providing no beneficial or detrimental role in the process. Previously, CD90.2 antibody
depletion has been expected to partially deplete NK cells in addition to ILCs, and thus depletion
of NK cells generally accompanies ILC depletion in order to rule out the effect of NK cell loss
on observed phenotypes (Eisenring et al., 2010;Monticelli et al., 2011).However, NK cell-
depleted Rag1-/-
animals were much more sensitive to MC38met-Luc expansion than their ILC-
depleted counterparts and Jak3W81R mice, with a trend towards higher lung tumour burdens and

87
metastatic coverage (Figure 14B, C). This suggests a dominant role for NK cell in controlling
MC38met-Luc lung expansion. While JakW81R
and Rag1-/-
mice treated with NK cell-depleting
antibody similarly do not contain NK cells, it might be expected that the presence of other
functional immune cells in the Rag1-/-
animals, such as ILCs might still protect these mice
compared to the Jak3-depleted animals. Thus, one might expect a less severe phenotype to be
observed in NK cell-depleted Rag1-/-
animals as opposed to the immune-deficient JakW81R
. This
was not the case however, and it is possible that the Jak3-lacking mice still contain immune
effectors which provide them with a greater degree of resistance to MC38met-Luc cell expansion
than NK cell-depleted Rag1-/-
mice. Another possibility for the unexpected hyper-susceptibility of
NK cell-depleted Rag1-/-
mice could be due to accidental targeting other non-NK cell immune
effectors in anti GM1-treated mice. However, as mentioned before, these results do not preclude
that ILCs may still play a role in the primary stages of CRC progression, and in fact, recent
studies have found a role for type III ILCs in promoting inflammation-driven CRC, through
production of Il-22 and Il-23 (Kirchberger et al., 2013;Chan et al., 2014).
Screening ENU mutant mice subjected to a disease models has allowed researchers to
draw links between genes and disease outcomes. For instance, ENU screening in mice has led to
the discovery of mice which were resistant to cerebral malaria, and which had a dominant
negative mutation in Jak3 (Bongfen et al., 2012). These findings highlighted the importance of
the detrimental effect of Jak3-dependent cells, such as CD8+ T cell in perpetuating the disease.
This unbiased approach thus has the potential to provide fresh insight into the area of cancer-host
interactions and thus also provides the possibility for the exploration of novel therapeutic
avenues in CRC intervention.

88
Using an ENU platform to screen for mutants with resistance or susceptibility to
MC38met-Luc colonization of the lung, our lab was able to identify two families which were
resistant to MC38met-Luc expansion. Interpreting resistance or susceptibility was initially
difficult in our screen, given the extreme variability of the MC38met-Luc cells in their in vivo
expansion, leading to variation of as much as 5 weeks in WT controls. While this variation was
later minimized using a strictly consistent cell culture schedule, it is important to consider the
variability in our metastatic CRC model when interpreting our results, especially given that
neither family contained a novel homozygous SNP across all 3 deviant animals analyzed by
exome sequencing. Generally however, mice were considered resistant if they survived longer
than WT and susceptible if they died alongside the Jak3W81R
mice. However, the most important
factor in determining a deviant family was the repetition of this observed phenotype, and the 1/4
segregation of this phenotype within the family. Thus, for instance, if all the mice in a family
died after WT mice, this was considered to be due to variation in the model. If however, we
observed one mouse out of four mice dying after WT, and this observation repeated itself in
subsequent injection rounds, this family was flagged as a potential deviant and candidate for
exome sequencing. Exome sequencing analysis led to a list of 2 candidate gene mutations in
Family 13 and 4 candidate gene mutations in Family 31.
Assuming that the candidate mutations are creating loss-of-function alleles, our lab could
use mice which are knock out for the candidate genes, or which have mutations for these genes,
in order to see if they recapitulate the resistant phenotype observed in our deviant animals
following MC38met-Luc injection and validate the metastasis resistance phenotype observed in
our deviant ENU-generated animals. However, it is a possibility that the mutations do not cause
reduced protein levels, and that they might disrupt, alter, or augment normal protein functions,

89
rendering knockout mice not an ideal model to study. For instance, if the mutation in question
actually increases the activity of the gene, a knock-in mouse model could be used to validate our
observations in the ENU mutants following MC38met-Luc injection.
Following genetic validation, assays such as immunohistochemistry, flow cytometry,
western blotting and immunoprecipitation could help determine protein expression, interacting
partners and function in controlling MC38met-Luc expansion in our resistant animals. It is useful
to next validate candidate genes in human tissues, for example to determine whether
decreased/increased expression of their encoded protein correlates with better or worse prognosis
in CRC patients. Achieving this step could aid in our understanding of CRC metastasis in
humans, potentially leading to a new prognostic marker for CRC patients, which could aid
physician in staging CRC patients or determining their treatment strategy, or could even lead to a
new therapeutic target for CRC.
CONCLUSION
Stephen Paget’s contribution to the understanding of cancer progression and pathogenesis
has been vital for the current development of cancer therapies. Since Hanahan and Weinberg’s
Hallmarks of Cancer, much research has focused on the contribution of the host
microenvironment for supporting the transformation of cells to a cancerous state, as well as their
subsequent invasion and metastases. The host environment has the potential to provide a
spectrum of factors which promote tumor growth, cancer cell survival, angiogenesis and organ
tropism (Langley and Fidler, 2011). Understanding this interplay between host and cancer has
helped physicians provide a more accurate prognosis for their patients, and has proven to be of
vital importance when creating therapeutic strategies for the disease. Similarly, gaining a

90
heightened understanding of the pre-metastatic niche will have important implications for the
treatment and staging of cancer in the coming years. The priming of a secondary site for
colonisation with tumor cells facilitates the establishment of secondary tumors. Keeping in mind
that metastasis is responsible for up to 90% of cancer-related deaths, and that current treatment
options at the metastatic stage focus on prolonging life rather than curing the disease, the pre-
metastatic niche seems to be an important focus in the quest for more effective metastatic-stage
therapies. Thus, preventing, targeting or even reversing the pre-metastatic niche could have
potent effects on the metastatic process, and has the potential to completely change the face of
stage IV cancer treatment as it is known today.
Lastly, the process of immunoediting has profound implications on the outcome of the
disease due to the plasticity of cancer cells and their ability to adapt to selective pressures while
simultaneously changing the environment to meet their needs. Understanding cancer
immunoediting and the impact of the immune system on the progression of cancer have aided in
the invention of a number of immune-based cancer therapies. However, challenges remain in
understanding how and why some tumors escape control, while others remain dormant.
Understanding host-specific factors and how they act to shape the pathogenesis of cancer can
lead to more targeted, effective treatments, while minimizing unnecessary side-effects of the
“one-size-fits-all” cancer treatment paradigm. Personalized medicine will allow healthcare
professionals to harness the specific aspects of the immune system to treat cancer patients in a
manner that more accurately reflects their needs based on their specific cancer sub-type. The
mechanisms behind tumor dormancy and the equilibrium stage of cancer are poorly understood
due to the unique challenges presented by attempting to model this state in mice. As such, most
of the acquired knowledge on the equilibrium stage comes from anecdotal evidence in humans

91
(Koebel et al., 2007;Teng et al., 2012). While studying this stage of the immunoediting process
remains a challenge, certain biomarkers, along with imaging technology, could allow for
expansion in this field, especially in the characterization of circulating tumor cells and their
niches. Most cancers are likely to be diagnosed during the equilibrium or escape stages, making
these two phases the prime target of current cancer treatments. Given the increased aggression
and ability for evasion that cancers often exhibit during the escape phase, the equilibrium phase
offers the potential to provide a period of time where the cancer may be particularly susceptible
to clinical intervention. Thus, research into further host factors influencing this stage could have
a powerful effect on curbing the devastating effects of cancer.
Cancer remains one of the primary causes of death in the developed world, and CRC in
particular presents a challenge even for the wealthy, modern healthcare system found in these
countries. While an accurate model of CRC metastasis remains elusive in mice, scientists have
nonetheless developed tools to more accurately understand the host and cancer factors which
contribute to cancer pathogenesis. As a more comprehensive understanding of CRC and its
metastasis emerges, there is hope that host mechanisms and understanding of the pre-metastatic
niche will provide new treatments. The management of metastasis in CRC, as well as other
cancers, at this stage only functions short-term, prolonging life rather than curing patients.
However, the potential for manipulation of host immune factors provides a promising start for
more effective metastatic-stage drugs, and if successful, these could radically change the face of
cancer care and the dismal prognoses for patients with metastatic cancer.

92
BIBLIOGRAPHY (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330-
337. (2013). NOD2 protects against colorectal cancer via regulation of gut bacteria. Cancer Discov 3, OF17. Aaltonen, L., Johns, L., Jarvinen, H., Mecklin, J.P., and Houlston, R. (2007). Explaining the familial
colorectal cancer risk associated with mismatch repair (MMR)-deficient and MMR-stable tumors. Clin Cancer Res 13, 356-361.
Ahearn, T.U., Shaukat, A., Flanders, W.D., Seabrook, M.E., and Bostick, R.M. (2012). Markers of the APC/beta-catenin signaling pathway as potential treatable, preneoplastic biomarkers of risk for colorectal neoplasms. Cancer Epidemiol Biomarkers Prev 21, 969-979.
Aihara, H., Kumar, N., and Thompson, C.C. (2014). Diagnosis, surveillance, and treatment strategies for familial adenomatous polyposis: rationale and update. Eur J Gastroenterol Hepatol 26, 255-262.
Allegra, C.J., Yothers, G., O'connell, M.J., Sharif, S., Petrelli, N.J., Colangelo, L.H., Atkins, J.N., Seay, T.E., Fehrenbacher, L., Goldberg, R.M., O'reilly, S., Chu, L., Azar, C.A., Lopa, S., and Wolmark, N. (2011). Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol 29, 11-16.
Almand, B., Resser, J.R., Lindman, B., Nadaf, S., Clark, J.I., Kwon, E.D., Carbone, D.P., and Gabrilovich, D.I. (2000). Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res 6, 1755-1766.
Amit, S., Hatzubai, A., Birman, Y., Andersen, J.S., Ben-Shushan, E., Mann, M., Ben-Neriah, Y., and Alkalay, I. (2002). Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev 16, 1066-1076.
Andreyev, H.J., Norman, A.R., Cunningham, D., Oates, J., Dix, B.R., Iacopetta, B.J., Young, J., Walsh, T., Ward, R., Hawkins, N., Beranek, M., Jandik, P., Benamouzig, R., Jullian, E., Laurent-Puig, P., Olschwang, S., Muller, O., Hoffmann, I., Rabes, H.M., Zietz, C., Troungos, C., Valavanis, C., Yuen, S.T., Ho, J.W., Croke, C.T., O'donoghue, D.P., Giaretti, W., Rapallo, A., Russo, A., Bazan, V., Tanaka, M., Omura, K., Azuma, T., Ohkusa, T., Fujimori, T., Ono, Y., Pauly, M., Faber, C., Glaesener, R., De Goeij, A.F., Arends, J.W., Andersen, S.N., Lovig, T., Breivik, J., Gaudernack, G., Clausen, O.P., De Angelis, P.D., Meling, G.I., Rognum, T.O., Smith, R., Goh, H.S., Font, A., Rosell, R., Sun, X.F., Zhang, H., Benhattar, J., Losi, L., Lee, J.Q., Wang, S.T., Clarke, P.A., Bell, S., Quirke, P., Bubb, V.J., Piris, J., Cruickshank, N.R., Morton, D., Fox, J.C., Al-Mulla, F., Lees, N., Hall, C.N., Snary, D., Wilkinson, K., Dillon, D., Costa, J., Pricolo, V.E., Finkelstein, S.D., Thebo, J.S., Senagore, A.J., Halter, S.A., Wadler, S., Malik, S., Krtolica, K., and Urosevic, N. (2001). Kirsten ras mutations in patients with colorectal cancer: the 'RASCAL II' study. Br J Cancer 85, 692-696.
Aretz, S., Stienen, D., Uhlhaas, S., Stolte, M., Entius, M.M., Loff, S., Back, W., Kaufmann, A., Keller, K.M., Blaas, S.H., Siebert, R., Vogt, S., Spranger, S., Holinski-Feder, E., Sunde, L., Propping, P., and Friedl, W. (2007). High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet 44, 702-709.
Asseman, C., Mauze, S., Leach, M.W., Coffman, R.L., and Powrie, F. (1999). An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J Exp Med 190, 995-1004.
Augustin, M., Sedlmeier, R., Peters, T., Huffstadt, U., Kochmann, E., Simon, D., Schoniger, M., Garke-Mayerthaler, S., Laufs, J., Mayhaus, M., Franke, S., Klose, M., Graupner, A., Kurzmann, M., Zinser, C., Wolf, A., Voelkel, M., Kellner, M., Kilian, M., Seelig, S., Koppius, A., Teubner, A., Korthaus, D., Nehls, M., and Wattler, S. (2005). Efficient and fast targeted production of murine models based on ENU mutagenesis. Mamm Genome 16, 405-413.

93
Balint, E.E., and Vousden, K.H. (2001). Activation and activities of the p53 tumour suppressor protein. Br J Cancer 85, 1813-1823.
Barnetson, R.A., Tenesa, A., Farrington, S.M., Nicholl, I.D., Cetnarskyj, R., Porteous, M.E., Campbell, H., and Dunlop, M.G. (2006). Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med 354, 2751-2763.
Barrett, J.C., Lee, J.C., Lees, C.W., Prescott, N.J., Anderson, C.A., Phillips, A., Wesley, E., Parnell, K., Zhang, H., Drummond, H., Nimmo, E.R., Massey, D., Blaszczyk, K., Elliott, T., Cotterill, L., Dallal, H., Lobo, A.J., Mowat, C., Sanderson, J.D., Jewell, D.P., Newman, W.G., Edwards, C., Ahmad, T., Mansfield, J.C., Satsangi, J., Parkes, M., Mathew, C.G., Donnelly, P., Peltonen, L., Blackwell, J.M., Bramon, E., Brown, M.A., Casas, J.P., Corvin, A., Craddock, N., Deloukas, P., Duncanson, A., Jankowski, J., Markus, H.S., Mccarthy, M.I., Palmer, C.N., Plomin, R., Rautanen, A., Sawcer, S.J., Samani, N., Trembath, R.C., Viswanathan, A.C., Wood, N., Spencer, C.C., Bellenguez, C., Davison, D., Freeman, C., Strange, A., Langford, C., Hunt, S.E., Edkins, S., Gwilliam, R., Blackburn, H., Bumpstead, S.J., Dronov, S., Gillman, M., Gray, E., Hammond, N., Jayakumar, A., Mccann, O.T., Liddle, J., Perez, M.L., Potter, S.C., Ravindrarajah, R., Ricketts, M., Waller, M., Weston, P., Widaa, S., Whittaker, P., Attwood, A.P., Stephens, J., Sambrook, J., Ouwehand, W.H., Mcardle, W.L., Ring, S.M., and Strachan, D.P. (2009). Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet 41, 1330-1334.
Becker, C., Fantini, M.C., Schramm, C., Lehr, H.A., Wirtz, S., Nikolaev, A., Burg, J., Strand, S., Kiesslich, R., Huber, S., Ito, H., Nishimoto, N., Yoshizaki, K., Kishimoto, T., Galle, P.R., Blessing, M., Rose-John, S., and Neurath, M.F. (2004). TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491-501.
Beggs, A.D., Latchford, A.R., Vasen, H.F., Moslein, G., Alonso, A., Aretz, S., Bertario, L., Blanco, I., Bulow, S., Burn, J., Capella, G., Colas, C., Friedl, W., Moller, P., Hes, F.J., Jarvinen, H., Mecklin, J.P., Nagengast, F.M., Parc, Y., Phillips, R.K., Hyer, W., Ponz De Leon, M., Renkonen-Sinisalo, L., Sampson, J.R., Stormorken, A., Tejpar, S., Thomas, H.J., Wijnen, J.T., Clark, S.K., and Hodgson, S.V. (2010). Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 59, 975-986.
Behrens, J., Von Kries, J.P., Kuhl, M., Bruhn, L., Wedlich, D., Grosschedl, R., and Birchmeier, W. (1996). Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 382, 638-642.
Bertrand, M.J., Doiron, K., Labbe, K., Korneluk, R.G., Barker, P.A., and Saleh, M. (2009). Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 30, 789-801.
Bevins, C.L., and Salzman, N.H. (2011). Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9, 356-368.
Bharadwaj, U., Li, M., Zhang, R., Chen, C., and Yao, Q. (2007). Elevated interleukin-6 and G-CSF in human pancreatic cancer cell conditioned medium suppress dendritic cell differentiation and activation. Cancer Res 67, 5479-5488.
Bhowmick, N.A., Chytil, A., Plieth, D., Gorska, A.E., Dumont, N., Shappell, S., Washington, M.K., Neilson, E.G., and Moses, H.L. (2004a). TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303, 848-851.
Bhowmick, N.A., Neilson, E.G., and Moses, H.L. (2004b). Stromal fibroblasts in cancer initiation and progression. Nature 432, 332-337.
Bilic, J., Huang, Y.L., Davidson, G., Zimmermann, T., Cruciat, C.M., Bienz, M., and Niehrs, C. (2007). Wnt induces LRP6 signalosomes and promotes dishevelled-dependent LRP6 phosphorylation. Science 316, 1619-1622.

94
Biswas, S., Chytil, A., Washington, K., Romero-Gallo, J., Gorska, A.E., Wirth, P.S., Gautam, S., Moses, H.L., and Grady, W.M. (2004). Transforming growth factor beta receptor type II inactivation promotes the establishment and progression of colon cancer. Cancer Res 64, 4687-4692.
Biswas, S., Trobridge, P., Romero-Gallo, J., Billheimer, D., Myeroff, L.L., Willson, J.K., Markowitz, S.D., and Grady, W.M. (2008). Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes Chromosomes Cancer 47, 95-106.
Bochkarev, A., Bochkareva, E., Frappier, L., and Edwards, A.M. (1999). The crystal structure of the complex of replication protein A subunits RPA32 and RPA14 reveals a mechanism for single-stranded DNA binding. EMBO J 18, 4498-4504.
Boehm, M., and Bonifacino, J.S. (2001). Adaptins: the final recount. Mol Biol Cell 12, 2907-2920. Boivin, G.P., Washington, K., Yang, K., Ward, J.M., Pretlow, T.P., Russell, R., Besselsen, D.G., Godfrey,
V.L., Doetschman, T., Dove, W.F., Pitot, H.C., Halberg, R.B., Itzkowitz, S.H., Groden, J., and Coffey, R.J. (2003). Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology 124, 762-777.
Boles, M.K., Wilkinson, B.M., Wilming, L.G., Liu, B., Probst, F.J., Harrow, J., Grafham, D., Hentges, K.E., Woodward, L.P., Maxwell, A., Mitchell, K., Risley, M.D., Johnson, R., Hirschi, K., Lupski, J.R., Funato, Y., Miki, H., Marin-Garcia, P., Matthews, L., Coffey, A.J., Parker, A., Hubbard, T.J., Rogers, J., Bradley, A., Adams, D.J., and Justice, M.J. (2009). Discovery of candidate disease genes in ENU-induced mouse mutants by large-scale sequencing, including a splice-site mutation in nucleoredoxin. PLoS Genet 5, e1000759.
Bolocan, A., Ion, D., Stoian, R.V., and Serban, M.B. (2011). Map syndrome (MYH Associated Polyposis) colorectal cancer, etiopathological connections. J Med Life 4, 109-111.
Bonaccorsi, I., Morandi, B., Antsiferova, O., Costa, G., Oliveri, D., Conte, R., Pezzino, G., Vermiglio, G., Anastasi, G.P., Navarra, G., Munz, C., Di Carlo, E., Mingari, M.C., and Ferlazzo, G. (2014). Membrane transfer from tumor cells overcomes deficient phagocytic ability of plasmacytoid dendritic cells for the acquisition and presentation of tumor antigens. J Immunol 192, 824-832.
Bongfen, S.E., Rodrigue-Gervais, I.G., Berghout, J., Torre, S., Cingolani, P., Wiltshire, S.A., Leiva-Torres, G.A., Letourneau, L., Sladek, R., Blanchette, M., Lathrop, M., Behr, M.A., Gruenheid, S., Vidal, S.M., Saleh, M., and Gros, P. (2012). An N-ethyl-N-nitrosourea (ENU)-induced dominant negative mutation in the JAK3 kinase protects against cerebral malaria. PLoS One 7, e31012.
Bos, J.L., Fearon, E.R., Hamilton, S.R., Verlaan-De Vries, M., Van Boom, J.H., Van Der Eb, A.J., and Vogelstein, B. (1987). Prevalence of ras gene mutations in human colorectal cancers. Nature 327, 293-297.
Bos, R., Van Der Groep, P., Greijer, A.E., Shvarts, A., Meijer, S., Pinedo, H.M., Semenza, G.L., Van Diest, P.J., and Van Der Wall, E. (2003). Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 97, 1573-1581.
Botma, A., Nagengast, F.M., Braem, M.G., Hendriks, J.C., Kleibeuker, J.H., Vasen, H.F., and Kampman, E. (2010). Body mass index increases risk of colorectal adenomas in men with Lynch syndrome: the GEOLynch cohort study. J Clin Oncol 28, 4346-4353.
Boyle, T., Keegel, T., Bull, F., Heyworth, J., and Fritschi, L. (2012). Physical activity and risks of proximal and distal colon cancers: a systematic review and meta-analysis. J Natl Cancer Inst 104, 1548-1561.
Brandl, K., Plitas, G., Schnabl, B., Dematteo, R.P., and Pamer, E.G. (2007). MyD88-mediated signals induce the bactericidal lectin RegIII gamma and protect mice against intestinal Listeria monocytogenes infection. J Exp Med 204, 1891-1900.

95
Brenner, H., Chang-Claude, J., Seiler, C.M., Rickert, A., and Hoffmeister, M. (2011). Protection from colorectal cancer after colonoscopy: a population-based, case-control study. Ann Intern Med 154, 22-30.
Brensinger, J.D., Laken, S.J., Luce, M.C., Powell, S.M., Vance, G.H., Ahnen, D.J., Petersen, G.M., Hamilton, S.R., and Giardiello, F.M. (1998). Variable phenotype of familial adenomatous polyposis in pedigrees with 3' mutation in the APC gene. Gut 43, 548-552.
Broderick, P., Carvajal-Carmona, L., Pittman, A.M., Webb, E., Howarth, K., Rowan, A., Lubbe, S., Spain, S., Sullivan, K., Fielding, S., Jaeger, E., Vijayakrishnan, J., Kemp, Z., Gorman, M., Chandler, I., Papaemmanuil, E., Penegar, S., Wood, W., Sellick, G., Qureshi, M., Teixeira, A., Domingo, E., Barclay, E., Martin, L., Sieber, O., Kerr, D., Gray, R., Peto, J., Cazier, J.B., Tomlinson, I., and Houlston, R.S. (2007). A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 39, 1315-1317.
Brosens, L.A., Langeveld, D., Van Hattem, W.A., Giardiello, F.M., and Offerhaus, G.J. (2011). Juvenile polyposis syndrome. World J Gastroenterol 17, 4839-4844.
Bulow, S., Faurschou Nielsen, T., Bulow, C., Bisgaard, M.L., Karlsen, L., and Moesgaard, F. (1996). The incidence rate of familial adenomatous polyposis. Results from the Danish Polyposis Register. Int J Colorectal Dis 11, 88-91.
Burn, J., Gerdes, A.M., Macrae, F., Mecklin, J.P., Moeslein, G., Olschwang, S., Eccles, D., Evans, D.G., Maher, E.R., Bertario, L., Bisgaard, M.L., Dunlop, M.G., Ho, J.W., Hodgson, S.V., Lindblom, A., Lubinski, J., Morrison, P.J., Murday, V., Ramesar, R., Side, L., Scott, R.J., Thomas, H.J., Vasen, H.F., Barker, G., Crawford, G., Elliott, F., Movahedi, M., Pylvanainen, K., Wijnen, J.T., Fodde, R., Lynch, H.T., Mathers, J.C., and Bishop, D.T. (2011). Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 378, 2081-2087.
Burnet, M. (1957). Cancer; a biological approach. I. The processes of control. Br Med J 1, 779-786. Carpentino, J.E., Hynes, M.J., Appelman, H.D., Zheng, T., Steindler, D.A., Scott, E.W., and Huang, E.H.
(2009). Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer Res 69, 8208-8215.
Chamaillard, M., Hashimoto, M., Horie, Y., Masumoto, J., Qiu, S., Saab, L., Ogura, Y., Kawasaki, A., Fukase, K., Kusumoto, S., Valvano, M.A., Foster, S.J., Mak, T.W., Nunez, G., and Inohara, N. (2003). An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 4, 702-707.
Chambers, A.F., Groom, A.C., and Macdonald, I.C. (2002). Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2, 563-572.
Chan, I.H., Jain, R., Tessmer, M.S., Gorman, D., Mangadu, R., Sathe, M., Vives, F., Moon, C., Penaflor, E., Turner, S., Ayanoglu, G., Chang, C., Basham, B., Mumm, J.B., Pierce, R.H., Yearley, J.H., Mcclanahan, T.K., Phillips, J.H., Cua, D.J., Bowman, E.P., Kastelein, R.A., and Laface, D. (2014). Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol 7, 842-856.
Chen, J., Lu, Y., Xu, J., Huang, Y., Cheng, H., Hu, G., Luo, C., Lou, M., Cao, G., Xie, Y., and Ying, K. (2004). Identification and characterization of NBEAL1, a novel human neurobeachin-like 1 protein gene from fetal brain, which is up regulated in glioma. Brain Res Mol Brain Res 125, 147-155.
Cheng, L., Huang, Z., Zhou, W., Wu, Q., Donnola, S., Liu, J.K., Fang, X., Sloan, A.E., Mao, Y., Lathia, J.D., Min, W., Mclendon, R.E., Rich, J.N., and Bao, S. (2013). Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153, 139-152.
Church, J. (2005). Ileoanal pouch neoplasia in familial adenomatous polyposis: an underestimated threat. Dis Colon Rectum 48, 1708-1713.

96
Clevers, H.C., and Bevins, C.L. (2013). Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol 75, 289-311.
Colditz, G.A., Atwood, K.A., Emmons, K., Monson, R.R., Willett, W.C., Trichopoulos, D., and Hunter, D.J. (2000). Harvard report on cancer prevention volume 4: Harvard Cancer Risk Index. Risk Index Working Group, Harvard Center for Cancer Prevention. Cancer Causes Control 11, 477-488.
Conte, D., Holcik, M., Lefebvre, C.A., Lacasse, E., Picketts, D.J., Wright, K.E., and Korneluk, R.G. (2006). Inhibitor of apoptosis protein cIAP2 is essential for lipopolysaccharide-induced macrophage survival. Mol Cell Biol 26, 699-708.
Corradetti, M.N., Inoki, K., Bardeesy, N., Depinho, R.A., and Guan, K.L. (2004). Regulation of the TSC pathway by LKB1: evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev 18, 1533-1538.
Corzo, C.A., Condamine, T., Lu, L., Cotter, M.J., Youn, J.I., Cheng, P., Cho, H.I., Celis, E., Quiceno, D.G., Padhya, T., Mccaffrey, T.V., Mccaffrey, J.C., and Gabrilovich, D.I. (2010). HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med 207, 2439-2453.
Costa, N.L., Valadares, M.C., Souza, P.P., Mendonca, E.F., Oliveira, J.C., Silva, T.A., and Batista, A.C. (2013). Tumor-associated macrophages and the profile of inflammatory cytokines in oral squamous cell carcinoma. Oral Oncol 49, 216-223.
Coussens, L.M., Raymond, W.W., Bergers, G., Laig-Webster, M., Behrendtsen, O., Werb, Z., Caughey, G.H., and Hanahan, D. (1999). Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev 13, 1382-1397.
Coussens, L.M., and Werb, Z. (2002). Inflammation and cancer. Nature 420, 860-867. Couturier-Maillard, A., Secher, T., Rehman, A., Normand, S., De Arcangelis, A., Haesler, R., Huot, L.,
Grandjean, T., Bressenot, A., Delanoye-Crespin, A., Gaillot, O., Schreiber, S., Lemoine, Y., Ryffel, B., Hot, D., Nunez, G., Chen, G., Rosenstiel, P., and Chamaillard, M. (2013). NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest 123, 700-711.
Crosnier, C., Stamataki, D., and Lewis, J. (2006). Organizing cell renewal in the intestine: stem cells, signals and combinatorial control. Nat Rev Genet 7, 349-359.
Cui, R., Okada, Y., Jang, S.G., Ku, J.L., Park, J.G., Kamatani, Y., Hosono, N., Tsunoda, T., Kumar, V., Tanikawa, C., Kamatani, N., Yamada, R., Kubo, M., Nakamura, Y., and Matsuda, K. (2011). Common variant in 6q26-q27 is associated with distal colon cancer in an Asian population. Gut 60, 799-805.
Dales, J.P., Garcia, S., Meunier-Carpentier, S., Andrac-Meyer, L., Haddad, O., Lavaut, M.N., Allasia, C., Bonnier, P., and Charpin, C. (2005). Overexpression of hypoxia-inducible factor HIF-1alpha predicts early relapse in breast cancer: retrospective study in a series of 745 patients. Int J Cancer 116, 734-739.
Damgaard, R.B., Nachbur, U., Yabal, M., Wong, W.W., Fiil, B.K., Kastirr, M., Rieser, E., Rickard, J.A., Bankovacki, A., Peschel, C., Ruland, J., Bekker-Jensen, S., Mailand, N., Kaufmann, T., Strasser, A., Walczak, H., Silke, J., Jost, P.J., and Gyrd-Hansen, M. (2012). The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 46, 746-758.
De Vos Tot Nederveen Cappel, W.H., Nagengast, F.M., Griffioen, G., Menko, F.H., Taal, B.G., Kleibeuker, J.H., and Vasen, H.F. (2002). Surveillance for hereditary nonpolyposis colorectal cancer: a long-term study on 114 families. Dis Colon Rectum 45, 1588-1594.
Delnatte, C., Sanlaville, D., Mougenot, J.F., Vermeesch, J.R., Houdayer, C., Blois, M.C., Genevieve, D., Goulet, O., Fryns, J.P., Jaubert, F., Vekemans, M., Lyonnet, S., Romana, S., Eng, C., and Stoppa-Lyonnet, D. (2006). Contiguous gene deletion within chromosome arm 10q is associated with

97
juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet 78, 1066-1074.
Derry, M.M., Raina, K., Agarwal, R., and Agarwal, C. (2014). Characterization of azoxymethane-induced colon tumor metastasis to lung in a mouse model relevant to human sporadic colorectal cancer and evaluation of grape seed extract efficacy. Exp Toxicol Pathol 66, 235-242.
Desai, T.K., and Barkel, D. (2008). Syndromic colon cancer: lynch syndrome and familial adenomatous polyposis. Gastroenterol Clin North Am 37, 47-72, vi.
Dewanji, A., Jeon, J., Meza, R., and Luebeck, E.G. (2011). Number and size distribution of colorectal adenomas under the multistage clonal expansion model of cancer. PLoS Comput Biol 7, e1002213.
Diergaarde, B., Braam, H., Vasen, H.F., Nagengast, F.M., Van Muijen, G.N., Kok, F.J., and Kampman, E. (2007). Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 5, 736-742.
Diez-Roux, G., Banfi, S., Sultan, M., Geffers, L., Anand, S., Rozado, D., Magen, A., Canidio, E., Pagani, M., Peluso, I., Lin-Marq, N., Koch, M., Bilio, M., Cantiello, I., Verde, R., De Masi, C., Bianchi, S.A., Cicchini, J., Perroud, E., Mehmeti, S., Dagand, E., Schrinner, S., Nurnberger, A., Schmidt, K., Metz, K., Zwingmann, C., Brieske, N., Springer, C., Hernandez, A.M., Herzog, S., Grabbe, F., Sieverding, C., Fischer, B., Schrader, K., Brockmeyer, M., Dettmer, S., Helbig, C., Alunni, V., Battaini, M.A., Mura, C., Henrichsen, C.N., Garcia-Lopez, R., Echevarria, D., Puelles, E., Garcia-Calero, E., Kruse, S., Uhr, M., Kauck, C., Feng, G., Milyaev, N., Ong, C.K., Kumar, L., Lam, M., Semple, C.A., Gyenesei, A., Mundlos, S., Radelof, U., Lehrach, H., Sarmientos, P., Reymond, A., Davidson, D.R., Dolle, P., Antonarakis, S.E., Yaspo, M.L., Martinez, S., Baldock, R.A., Eichele, G., and Ballabio, A. (2011). A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PLoS Biol 9, e1000582.
Dong, X., Xu, F., Gong, Y., Gao, J., Lin, P., Chen, T., Peng, Y., Qiang, B., Yuan, J., Peng, X., and Rao, Z. (2006). Crystal structure of the V domain of human Nectin-like molecule-1/Syncam3/Tsll1/Igsf4b, a neural tissue-specific immunoglobulin-like cell-cell adhesion molecule. J Biol Chem 281, 10610-10617.
Duffy, M.J., Lamerz, R., Haglund, C., Nicolini, A., Kalousova, M., Holubec, L., and Sturgeon, C. (2014). Tumor markers in colorectal cancer, gastric cancer and gastrointestinal stromal cancers: European group on tumor markers 2014 guidelines update. Int J Cancer 134, 2513-2522.
Dunlop, M.G., Dobbins, S.E., Farrington, S.M., Jones, A.M., Palles, C., Whiffin, N., Tenesa, A., Spain, S., Broderick, P., Ooi, L.Y., Domingo, E., Smillie, C., Henrion, M., Frampton, M., Martin, L., Grimes, G., Gorman, M., Semple, C., Ma, Y.P., Barclay, E., Prendergast, J., Cazier, J.B., Olver, B., Penegar, S., Lubbe, S., Chander, I., Carvajal-Carmona, L.G., Ballereau, S., Lloyd, A., Vijayakrishnan, J., Zgaga, L., Rudan, I., Theodoratou, E., Starr, J.M., Deary, I., Kirac, I., Kovacevic, D., Aaltonen, L.A., Renkonen-Sinisalo, L., Mecklin, J.P., Matsuda, K., Nakamura, Y., Okada, Y., Gallinger, S., Duggan, D.J., Conti, D., Newcomb, P., Hopper, J., Jenkins, M.A., Schumacher, F., Casey, G., Easton, D., Shah, M., Pharoah, P., Lindblom, A., Liu, T., Smith, C.G., West, H., Cheadle, J.P., Midgley, R., Kerr, D.J., Campbell, H., Tomlinson, I.P., and Houlston, R.S. (2012). Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet 44, 770-776.
Dunn, G.P., Koebel, C.M., and Schreiber, R.D. (2006). Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6, 836-848.
Dunn, G.P., Old, L.J., and Schreiber, R.D. (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21, 137-148.
Ebos, J.M., Lee, C.R., Cruz-Munoz, W., Bjarnason, G.A., Christensen, J.G., and Kerbel, R.S. (2009). Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232-239.

98
Edin, S., Wikberg, M.L., Rutegard, J., Oldenborg, P.A., and Palmqvist, R. (2013). Phenotypic skewing of macrophages in vitro by secreted factors from colorectal cancer cells. PLoS One 8, e74982.
Eisenring, M., Vom Berg, J., Kristiansen, G., Saller, E., and Becher, B. (2010). IL-12 initiates tumor rejection via lymphoid tissue-inducer cells bearing the natural cytotoxicity receptor NKp46. Nat Immunol 11, 1030-1038.
Erler, J.T., Bennewith, K.L., Cox, T.R., Lang, G., Bird, D., Koong, A., Le, Q.T., and Giaccia, A.J. (2009). Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35-44.
Fedirko, V., Tramacere, I., Bagnardi, V., Rota, M., Scotti, L., Islami, F., Negri, E., Straif, K., Romieu, I., La Vecchia, C., Boffetta, P., and Jenab, M. (2011). Alcohol drinking and colorectal cancer risk: an overall and dose-response meta-analysis of published studies. Ann Oncol 22, 1958-1972.
Ferlay, J., Shin, H.R., Bray, F., Forman, D., Mathers, C., and Parkin, D.M. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127, 2893-2917.
Fernandez-Rozadilla, C., Cazier, J.B., Tomlinson, I.P., Carvajal-Carmona, L.G., Palles, C., Lamas, M.J., Baiget, M., Lopez-Fernandez, L.A., Brea-Fernandez, A., Abuli, A., Bujanda, L., Clofent, J., Gonzalez, D., Xicola, R., Andreu, M., Bessa, X., Jover, R., Llor, X., Moreno, V., Castells, A., Carracedo, A., Castellvi-Bel, S., and Ruiz-Ponte, C. (2013). A colorectal cancer genome-wide association study in a Spanish cohort identifies two variants associated with colorectal cancer risk at 1p33 and 8p12. BMC Genomics 14, 55.
Fiaschi, T., Marini, A., Giannoni, E., Taddei, M.L., Gandellini, P., De Donatis, A., Lanciotti, M., Serni, S., Cirri, P., and Chiarugi, P. (2012). Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res 72, 5130-5140.
Filipazzi, P., Huber, V., and Rivoltini, L. (2012). Phenotype, function and clinical implications of myeloid-derived suppressor cells in cancer patients. Cancer Immunol Immunother 61, 255-263.
Fleming, N.I., Jorissen, R.N., Mouradov, D., Christie, M., Sakthianandeswaren, A., Palmieri, M., Day, F., Li, S., Tsui, C., Lipton, L., Desai, J., Jones, I.T., Mclaughlin, S., Ward, R.L., Hawkins, N.J., Ruszkiewicz, A.R., Moore, J., Zhu, H.J., Mariadason, J.M., Burgess, A.W., Busam, D., Zhao, Q., Strausberg, R.L., Gibbs, P., and Sieber, O.M. (2013). SMAD2, SMAD3 and SMAD4 mutations in colorectal cancer. Cancer Res 73, 725-735.
Forssell, J., Oberg, A., Henriksson, M.L., Stenling, R., Jung, A., and Palmqvist, R. (2007). High macrophage infiltration along the tumor front correlates with improved survival in colon cancer. Clin Cancer Res 13, 1472-1479.
Fukuhara, H., Kuramochi, M., Nobukuni, T., Fukami, T., Saino, M., Maruyama, T., Nomura, S., Sekiya, T., and Murakami, Y. (2001). Isolation of the TSLL1 and TSLL2 genes, members of the tumor suppressor TSLC1 gene family encoding transmembrane proteins. Oncogene 20, 5401-5407.
Gabitass, R.F., Annels, N.E., Stocken, D.D., Pandha, H.A., and Middleton, G.W. (2011). Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother 60, 1419-1430.
Gajewski, T.F., Fuertes, M., Spaapen, R., Zheng, Y., and Kline, J. (2011). Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol 23, 286-292.
Galizia, G., Gemei, M., Del Vecchio, L., Zamboli, A., Di Noto, R., Mirabelli, P., Salvatore, F., Castellano, P., Orditura, M., De Vita, F., Pinto, M., Pignatelli, C., and Lieto, E. (2012). Combined CD133/CD44 expression as a prognostic indicator of disease-free survival in patients with colorectal cancer. Arch Surg 147, 18-24.
Gallo, R.L., and Hooper, L.V. (2012). Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol 12, 503-516.

99
Gao, J., Chen, T., Liu, J., Liu, W., Hu, G., Guo, X., Yin, B., Gong, Y., Zhao, J., Qiang, B., Yuan, J., and Peng, X. (2009). Loss of NECL1, a novel tumor suppressor, can be restored in glioma by HDAC inhibitor-Trichostatin A through Sp1 binding site. Glia 57, 989-999.
Garrett, W.S., Gordon, J.I., and Glimcher, L.H. (2010). Homeostasis and inflammation in the intestine. Cell 140, 859-870.
Ghiringhelli, F., Puig, P.E., Roux, S., Parcellier, A., Schmitt, E., Solary, E., Kroemer, G., Martin, F., Chauffert, B., and Zitvogel, L. (2005). Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med 202, 919-929.
Gilsing, A.M., Berndt, S.I., Ruder, E.H., Graubard, B.I., Ferrucci, L.M., Burdett, L., Weissfeld, J.L., Cross, A.J., and Sinha, R. (2012). Meat-related mutagen exposure, xenobiotic metabolizing gene polymorphisms and the risk of advanced colorectal adenoma and cancer. Carcinogenesis 33, 1332-1339.
Granot, Z., Henke, E., Comen, E.A., King, T.A., Norton, L., and Benezra, R. (2011). Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell 20, 300-314.
Gridelli, C., Rossi, A., Maione, P., Rossi, E., Castaldo, V., Sacco, P.C., and Colantuoni, G. (2009). Vascular disrupting agents: a novel mechanism of action in the battle against non-small cell lung cancer. Oncologist 14, 612-620.
Grimm, M., Kim, M., Rosenwald, A., Von Raden, B.H., Tsaur, I., Meier, E., Heemann, U., Germer, C.T., Gasser, M., and Waaga-Gasser, A.M. (2010). Tumour-mediated TRAIL-Receptor expression indicates effective apoptotic depletion of infiltrating CD8+ immune cells in clinical colorectal cancer. Eur J Cancer 46, 2314-2323.
Grizzle, W.E., Manne, U., Jhala, N.C., and Weiss, H.L. (2001). Molecular characterization of colorectal neoplasia in translational research. Arch Pathol Lab Med 125, 91-98.
Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L., Albertsen, H., Joslyn, G., Stevens, J., Spirio, L., Robertson, M., and Et Al. (1991). Identification and characterization of the familial adenomatous polyposis coli gene. Cell 66, 589-600.
Grosso, J.F., and Jure-Kunkel, M.N. (2013). CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun 13, 5.
Guido, C., Whitaker-Menezes, D., Lin, Z., Pestell, R.G., Howell, A., Zimmers, T.A., Casimiro, M.C., Aquila, S., Ando, S., Martinez-Outschoorn, U.E., Sotgia, F., and Lisanti, M.P. (2012). Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 3, 798-810.
Gulubova, M.V., Ananiev, J.R., Vlaykova, T.I., Yovchev, Y., Tsoneva, V., and Manolova, I.M. (2012). Role of dendritic cells in progression and clinical outcome of colon cancer. Int J Colorectal Dis 27, 159-169.
Haanstra, J.F., De Vos Tot Nederveen Cappel, W.H., Gopie, J.P., Vecht, J., Vanhoutvin, S.A., Cats, A., Van Der Zaag-Loonen, H.J., Langers, A.M., Bergmann, J.H., Van De Meeberg, P.C., Dekker, E., Kleibeuker, J.H., Vasen, H.F., Nagengast, F.M., and Van Duijvendijk, P. (2012). Quality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomy. Dis Colon Rectum 55, 653-659.
Hadano, S., Hand, C.K., Osuga, H., Yanagisawa, Y., Otomo, A., Devon, R.S., Miyamoto, N., Showguchi-Miyata, J., Okada, Y., Singaraja, R., Figlewicz, D.A., Kwiatkowski, T., Hosler, B.A., Sagie, T., Skaug, J., Nasir, J., Brown, R.H., Jr., Scherer, S.W., Rouleau, G.A., Hayden, M.R., and Ikeda, J.E. (2001). A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet 29, 166-173.

100
Halford, S.E., Rowan, A.J., Lipton, L., Sieber, O.M., Pack, K., Thomas, H.J., Hodgson, S.V., Bodmer, W.F., and Tomlinson, I.P. (2003). Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol 162, 1545-1548.
Hammoud, S.S., Cairns, B.R., and Jones, D.A. (2013). Epigenetic regulation of colon cancer and intestinal stem cells. Curr Opin Cell Biol 25, 177-183.
Han, K.S., Kang, H.J., Kim, E.Y., Yoon, W.J., Sohn, S., Kwon, H.J., and Gwag, B.J. (2001). 1,2-bis(2-Aminophenoxy)ethane-N,N,N',N'-tetraacetic acid induces caspase-mediated apoptosis and reactive oxygen species-mediated necrosis in cultured cortical neurons. J Neurochem 78, 230-239.
Han, Y., Chen, Z., Yang, Y., Jiang, Z., Gu, Y., Liu, Y., Lin, C., Pan, Z., Yu, Y., Jiang, M., Zhou, W., and Cao, X. (2014). Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 59, 567-579.
Hanahan, D., and Weinberg, R.A. (2000). The hallmarks of cancer. Cell 100, 57-70. Hanahan, D., and Weinberg, R.A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646-674. Hansen, R.D., Albieri, V., Tjonneland, A., Overvad, K., Andersen, K.K., and Raaschou-Nielsen, O. (2013).
Effects of smoking and antioxidant micronutrients on risk of colorectal cancer. Clin Gastroenterol Hepatol 11, 406-415 e403.
Hartmann-Stuhler, C., and Prange, R. (2001). Hepatitis B virus large envelope protein interacts with gamma2-adaptin, a clathrin adaptor-related protein. J Virol 75, 5343-5351.
Hasegawa, M., Fujimoto, Y., Lucas, P.C., Nakano, H., Fukase, K., Nunez, G., and Inohara, N. (2008). A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J 27, 373-383.
Hasegawa, M., Yang, K., Hashimoto, M., Park, J.H., Kim, Y.G., Fujimoto, Y., Nunez, G., Fukase, K., and Inohara, N. (2006). Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem 281, 29054-29063.
He, X., Semenov, M., Tamai, K., and Zeng, X. (2004). LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development 131, 1663-1677.
Hearle, N., Schumacher, V., Menko, F.H., Olschwang, S., Boardman, L.A., Gille, J.J., Keller, J.J., Westerman, A.M., Scott, R.J., Lim, W., Trimbath, J.D., Giardiello, F.M., Gruber, S.B., Offerhaus, G.J., De Rooij, F.W., Wilson, J.H., Hansmann, A., Moslein, G., Royer-Pokora, B., Vogel, T., Phillips, R.K., Spigelman, A.D., and Houlston, R.S. (2006). Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 12, 3209-3215.
Hemminki, A., Markie, D., Tomlinson, I., Avizienyte, E., Roth, S., Loukola, A., Bignell, G., Warren, W., Aminoff, M., Hoglund, P., Jarvinen, H., Kristo, P., Pelin, K., Ridanpaa, M., Salovaara, R., Toro, T., Bodmer, W., Olschwang, S., Olsen, A.S., Stratton, M.R., De La Chapelle, A., and Aaltonen, L.A. (1998). A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391, 184-187.
Heng, T.S., and Painter, M.W. (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 9, 1091-1094.
Herbst, A., Jurinovic, V., Krebs, S., Thieme, S.E., Blum, H., Goke, B., and Kolligs, F.T. (2014). Comprehensive analysis of beta-catenin target genes in colorectal carcinoma cell lines with deregulated Wnt/beta-catenin signaling. BMC Genomics 15, 74.
Hiratsuka, S., Watanabe, A., Aburatani, H., and Maru, Y. (2006). Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8, 1369-1375.

101
Hiura, M., Hashimura, T., Watanabe, Y., Kuribayashi, K., and Yoshida, O. (1994). Induction of specific anti-tumour immunity by interferon-gamma gene-transferred murine bladder carcinoma MBT-2. Folia Biol (Praha) 40, 49-61.
Hosogi, H., Nagayama, S., Kawamura, J., Koshiba, Y., Nomura, A., Itami, A., Okabe, H., Satoh, S., Watanabe, G., and Sakai, Y. (2008). Molecular insights into Peutz-Jeghers syndrome: two probands with a germline mutation of LKB1. J Gastroenterol 43, 492-497.
Houghton, A.M., Rzymkiewicz, D.M., Ji, H., Gregory, A.D., Egea, E.E., Metz, H.E., Stolz, D.B., Land, S.R., Marconcini, L.A., Kliment, C.R., Jenkins, K.M., Beaulieu, K.A., Mouded, M., Frank, S.J., Wong, K.K., and Shapiro, S.D. (2010). Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med 16, 219-223.
Houlston, R.S., Cheadle, J., Dobbins, S.E., Tenesa, A., Jones, A.M., Howarth, K., Spain, S.L., Broderick, P., Domingo, E., Farrington, S., Prendergast, J.G., Pittman, A.M., Theodoratou, E., Smith, C.G., Olver, B., Walther, A., Barnetson, R.A., Churchman, M., Jaeger, E.E., Penegar, S., Barclay, E., Martin, L., Gorman, M., Mager, R., Johnstone, E., Midgley, R., Niittymaki, I., Tuupanen, S., Colley, J., Idziaszczyk, S., Thomas, H.J., Lucassen, A.M., Evans, D.G., Maher, E.R., Maughan, T., Dimas, A., Dermitzakis, E., Cazier, J.B., Aaltonen, L.A., Pharoah, P., Kerr, D.J., Carvajal-Carmona, L.G., Campbell, H., Dunlop, M.G., and Tomlinson, I.P. (2010). Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet 42, 973-977.
Houlston, R.S., Webb, E., Broderick, P., Pittman, A.M., Di Bernardo, M.C., Lubbe, S., Chandler, I., Vijayakrishnan, J., Sullivan, K., Penegar, S., Carvajal-Carmona, L., Howarth, K., Jaeger, E., Spain, S.L., Walther, A., Barclay, E., Martin, L., Gorman, M., Domingo, E., Teixeira, A.S., Kerr, D., Cazier, J.B., Niittymaki, I., Tuupanen, S., Karhu, A., Aaltonen, L.A., Tomlinson, I.P., Farrington, S.M., Tenesa, A., Prendergast, J.G., Barnetson, R.A., Cetnarskyj, R., Porteous, M.E., Pharoah, P.D., Koessler, T., Hampe, J., Buch, S., Schafmayer, C., Tepel, J., Schreiber, S., Volzke, H., Chang-Claude, J., Hoffmeister, M., Brenner, H., Zanke, B.W., Montpetit, A., Hudson, T.J., Gallinger, S., Campbell, H., and Dunlop, M.G. (2008). Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet 40, 1426-1435.
Hsu, L.C., Ali, S.R., Mcgillivray, S., Tseng, P.H., Mariathasan, S., Humke, E.W., Eckmann, L., Powell, J.J., Nizet, V., Dixit, V.M., and Karin, M. (2008). A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc Natl Acad Sci U S A 105, 7803-7808.
Huang, B., Lei, Z., Zhao, J., Gong, W., Liu, J., Chen, Z., Liu, Y., Li, D., Yuan, Y., Zhang, G.M., and Feng, Z.H. (2007). CCL2/CCR2 pathway mediates recruitment of myeloid suppressor cells to cancers. Cancer Lett 252, 86-92.
Huang, B., Pan, P.Y., Li, Q., Sato, A.I., Levy, D.E., Bromberg, J., Divino, C.M., and Chen, S.H. (2006). Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66, 1123-1131.
Huber, O., Korn, R., Mclaughlin, J., Ohsugi, M., Herrmann, B.G., and Kemler, R. (1996). Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev 59, 3-10.
Huh, J.W., Oh, B.R., Kim, H.R., and Kim, Y.J. (2010). Preoperative carcinoembryonic antigen level as an independent prognostic factor in potentially curative colon cancer. J Surg Oncol 101, 396-400.
Hung, K.E., Maricevich, M.A., Richard, L.G., Chen, W.Y., Richardson, M.P., Kunin, A., Bronson, R.T., Mahmood, U., and Kucherlapati, R. (2010). Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc Natl Acad Sci U S A 107, 1565-1570.

102
Hurlstone, D.P., Saunders, B.P., and Church, J.M. (2008). Endoscopic surveillance of the ileoanal pouch following restorative proctocolectomy for familial adenomatous polyposis. Endoscopy 40, 437-442.
Iacobuzio-Donahue, C.A., Song, J., Parmiagiani, G., Yeo, C.J., Hruban, R.H., and Kern, S.E. (2004). Missense mutations of MADH4: characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res 10, 1597-1604.
Ibrahim, A., Barnes, D.R., Dunlop, J., Barrowdale, D., Antoniou, A.C., and Berg, J.N. (2014). Attenuated familial adenomatous polyposis manifests as autosomal dominant late-onset colorectal cancer. Eur J Hum Genet.
Ikeda, F., Deribe, Y.L., Skanland, S.S., Stieglitz, B., Grabbe, C., Franz-Wachtel, M., Van Wijk, S.J., Goswami, P., Nagy, V., Terzic, J., Tokunaga, F., Androulidaki, A., Nakagawa, T., Pasparakis, M., Iwai, K., Sundberg, J.P., Schaefer, L., Rittinger, K., Macek, B., and Dikic, I. (2011). SHARPIN forms a linear ubiquitin ligase complex regulating NF-kappaB activity and apoptosis. Nature 471, 637-641.
Inohara, N., Koseki, T., Del Peso, L., Hu, Y., Yee, C., Chen, S., Carrio, R., Merino, J., Liu, D., Ni, J., and Nunez, G. (1999). Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J Biol Chem 274, 14560-14567.
Janssen, K.P., Alberici, P., Fsihi, H., Gaspar, C., Breukel, C., Franken, P., Rosty, C., Abal, M., El Marjou, F., Smits, R., Louvard, D., Fodde, R., and Robine, S. (2006). APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 131, 1096-1109.
Jarvinen, H.J., Aarnio, M., Mustonen, H., Aktan-Collan, K., Aaltonen, L.A., Peltomaki, P., De La Chapelle, A., and Mecklin, J.P. (2000). Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 118, 829-834.
Jasperson, K.W., Tuohy, T.M., Neklason, D.W., and Burt, R.W. (2010). Hereditary and familial colon cancer. Gastroenterology 138, 2044-2058.
Jenne, D.E., Reimann, H., Nezu, J., Friedel, W., Loff, S., Jeschke, R., Muller, O., Back, W., and Zimmer, M. (1998). Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18, 38-43.
Jia, W.H., Zhang, B., Matsuo, K., Shin, A., Xiang, Y.B., Jee, S.H., Kim, D.H., Ren, Z., Cai, Q., Long, J., Shi, J., Wen, W., Yang, G., Delahanty, R.J., Ji, B.T., Pan, Z.Z., Matsuda, F., Gao, Y.T., Oh, J.H., Ahn, Y.O., Park, E.J., Li, H.L., Park, J.W., Jo, J., Jeong, J.Y., Hosono, S., Casey, G., Peters, U., Shu, X.O., Zeng, Y.X., and Zheng, W. (2013). Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet 45, 191-196.
Jiao, S., Hsu, L., Berndt, S., Bezieau, S., Brenner, H., Buchanan, D., Caan, B.J., Campbell, P.T., Carlson, C.S., Casey, G., Chan, A.T., Chang-Claude, J., Chanock, S., Conti, D.V., Curtis, K.R., Duggan, D., Gallinger, S., Gruber, S.B., Harrison, T.A., Hayes, R.B., Henderson, B.E., Hoffmeister, M., Hopper, J.L., Hudson, T.J., Hutter, C.M., Jackson, R.D., Jenkins, M.A., Kantor, E.D., Kolonel, L.N., Kury, S., Le Marchand, L., Lemire, M., Newcomb, P.A., Potter, J.D., Qu, C., Rosse, S.A., Schoen, R.E., Schumacher, F.R., Seminara, D., Slattery, M.L., Ulrich, C.M., Zanke, B.W., and Peters, U. (2012). Genome-wide search for gene-gene interactions in colorectal cancer. PLoS One 7, e52535.
Jin, Y.R., and Yoon, J.K. (2012). The R-spondin family of proteins: emerging regulators of WNT signaling. Int J Biochem Cell Biol 44, 2278-2287.
Johansson-Lindbom, B., Svensson, M., Pabst, O., Palmqvist, C., Marquez, G., Forster, R., and Agace, W.W. (2005). Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med 202, 1063-1073.
Johansson, M.E., Phillipson, M., Petersson, J., Velcich, A., Holm, L., and Hansson, G.C. (2008). The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105, 15064-15069.

103
Jones, S., Chen, W.D., Parmigiani, G., Diehl, F., Beerenwinkel, N., Antal, T., Traulsen, A., Nowak, M.A., Siegel, C., Velculescu, V.E., Kinzler, K.W., Vogelstein, B., Willis, J., and Markowitz, S.D. (2008). Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A 105, 4283-4288.
Jotzu, C., Alt, E., Welte, G., Li, J., Hennessy, B.T., Devarajan, E., Krishnappa, S., Pinilla, S., Droll, L., and Song, Y.H. (2010). Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Anal Cell Pathol (Amst) 33, 61-79.
Joyce, J.A., and Pollard, J.W. (2009). Microenvironmental regulation of metastasis. Nat Rev Cancer 9, 239-252.
Kanth, V.V., Bhalsing, S., Sasikala, M., Rao, G.V., Pradeep, R., Avanthi, U.S., and Reddy, D.N. (2014). Microsatellite instability and promoter hypermethylation in colorectal cancer in India. Tumour Biol 35, 4347-4355.
Kaplan, D.H., Shankaran, V., Dighe, A.S., Stockert, E., Aguet, M., Old, L.J., and Schreiber, R.D. (1998). Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A 95, 7556-7561.
Kaplan, R.N., Riba, R.D., Zacharoulis, S., Bramley, A.H., Vincent, L., Costa, C., Macdonald, D.D., Jin, D.K., Shido, K., Kerns, S.A., Zhu, Z., Hicklin, D., Wu, Y., Port, J.L., Altorki, N., Port, E.R., Ruggero, D., Shmelkov, S.V., Jensen, K.K., Rafii, S., and Lyden, D. (2005). VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820-827.
Kartheuser, A., Stangherlin, P., Brandt, D., Remue, C., and Sempoux, C. (2006). Restorative proctocolectomy and ileal pouch-anal anastomosis for familial adenomatous polyposis revisited. Fam Cancer 5, 241-260; discussion 261-242.
Karuman, P., Gozani, O., Odze, R.D., Zhou, X.C., Zhu, H., Shaw, R., Brien, T.P., Bozzuto, C.D., Ooi, D., Cantley, L.C., and Yuan, J. (2001). The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell 7, 1307-1319.
Kato, I., Boleij, A., Kortman, G.A., Roelofs, R., Djuric, Z., Severson, R.K., and Tjalsma, H. (2013). Partial associations of dietary iron, smoking and intestinal bacteria with colorectal cancer risk. Nutr Cancer 65, 169-177.
Kempers, M.J., Kuiper, R.P., Ockeloen, C.W., Chappuis, P.O., Hutter, P., Rahner, N., Schackert, H.K., Steinke, V., Holinski-Feder, E., Morak, M., Kloor, M., Buttner, R., Verwiel, E.T., Van Krieken, J.H., Nagtegaal, I.D., Goossens, M., Van Der Post, R.S., Niessen, R.C., Sijmons, R.H., Kluijt, I., Hogervorst, F.B., Leter, E.M., Gille, J.J., Aalfs, C.M., Redeker, E.J., Hes, F.J., Tops, C.M., Van Nesselrooij, B.P., Van Gijn, M.E., Gomez Garcia, E.B., Eccles, D.M., Bunyan, D.J., Syngal, S., Stoffel, E.M., Culver, J.O., Palomares, M.R., Graham, T., Velsher, L., Papp, J., Olah, E., Chan, T.L., Leung, S.Y., Van Kessel, A.G., Kiemeney, L.A., Hoogerbrugge, N., and Ligtenberg, M.J. (2011). Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol 12, 49-55.
Kim, C.J., Cho, Y.G., Park, C.H., Kim, S.Y., Nam, S.W., Lee, S.H., Yoo, N.J., Lee, J.Y., and Park, W.S. (2004). Genetic alterations of the MYH gene in gastric cancer. Oncogene 23, 6820-6822.
Kim, S., Takahashi, H., Lin, W.W., Descargues, P., Grivennikov, S., Kim, Y., Luo, J.L., and Karin, M. (2009). Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 457, 102-106.
Kim, Y.S., and Ho, S.B. (2010). Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 12, 319-330.
Kinzler, K.W., Nilbert, M.C., Su, L.K., Vogelstein, B., Bryan, T.M., Levy, D.B., Smith, K.J., Preisinger, A.C., Hedge, P., Mckechnie, D., and Et Al. (1991). Identification of FAP locus genes from chromosome 5q21. Science 253, 661-665.

104
Kirchberger, S., Royston, D.J., Boulard, O., Thornton, E., Franchini, F., Szabady, R.L., Harrison, O., and Powrie, F. (2013). Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J Exp Med 210, 917-931.
Kobayashi, K., Inohara, N., Hernandez, L.D., Galan, J.E., Nunez, G., Janeway, C.A., Medzhitov, R., and Flavell, R.A. (2002). RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 416, 194-199.
Kobayashi, K.S., Chamaillard, M., Ogura, Y., Henegariu, O., Inohara, N., Nunez, G., and Flavell, R.A. (2005). Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731-734.
Koebel, C.M., Vermi, W., Swann, J.B., Zerafa, N., Rodig, S.J., Old, L.J., Smyth, M.J., and Schreiber, R.D. (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450, 903-907.
Kosmaczewska, A., Bocko, D., Ciszak, L., Wlodarska-Polinska, I., Kornafel, J., Szteblich, A., Masternak, A., and Frydecka, I. (2012). Dysregulated expression of both the costimulatory CD28 and inhibitory CTLA-4 molecules in PB T cells of advanced cervical cancer patients suggests systemic immunosuppression related to disease progression. Pathol Oncol Res 18, 479-489.
Kovacs, M.E., Papp, J., Szentirmay, Z., Otto, S., and Olah, E. (2009). Deletions removing the last exon of TACSTD1 constitute a distinct class of mutations predisposing to Lynch syndrome. Hum Mutat 30, 197-203.
Kowanetz, M., Wu, X., Lee, J., Tan, M., Hagenbeek, T., Qu, X., Yu, L., Ross, J., Korsisaari, N., Cao, T., Bou-Reslan, H., Kallop, D., Weimer, R., Ludlam, M.J., Kaminker, J.S., Modrusan, Z., Van Bruggen, N., Peale, F.V., Carano, R., Meng, Y.G., and Ferrara, N. (2010). Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci U S A 107, 21248-21255.
Koyama, M., Ito, M., Nagai, H., Emi, M., and Moriyama, Y. (1999). Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: identification of seven novel somatic mutations in tumors from Japanese patients. Mutat Res 406, 71-77.
Kraus, C., Liehr, T., Hulsken, J., Behrens, J., Birchmeier, W., Grzeschik, K.H., and Ballhausen, W.G. (1994). Localization of the human beta-catenin gene (CTNNB1) to 3p21: a region implicated in tumor development. Genomics 23, 272-274.
Krieg, A., Correa, R.G., Garrison, J.B., Le Negrate, G., Welsh, K., Huang, Z., Knoefel, W.T., and Reed, J.C. (2009). XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A 106, 14524-14529.
Kuida, K., Lippke, J.A., Ku, G., Harding, M.W., Livingston, D.J., Su, M.S., and Flavell, R.A. (1995). Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267, 2000-2003.
Kuperwasser, C., Chavarria, T., Wu, M., Magrane, G., Gray, J.W., Carey, L., Richardson, A., and Weinberg, R.A. (2004). Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Natl Acad Sci U S A 101, 4966-4971.
Labbe, K., Miu, J., Yeretssian, G., Serghides, L., Tam, M., Finney, C.A., Erdman, L.K., Goulet, M.L., Kain, K.C., Stevenson, M.M., and Saleh, M. (2010). Caspase-12 dampens the immune response to malaria independently of the inflammasome by targeting NF-kappaB signaling. J Immunol 185, 5495-5502.
Langley, R.R., and Fidler, I.J. (2011). The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer 128, 2527-2535.
Lascorz, J., Forsti, A., Chen, B., Buch, S., Steinke, V., Rahner, N., Holinski-Feder, E., Morak, M., Schackert, H.K., Gorgens, H., Schulmann, K., Goecke, T., Kloor, M., Engel, C., Buttner, R., Kunkel, N., Weires, M., Hoffmeister, M., Pardini, B., Naccarati, A., Vodickova, L., Novotny, J., Schreiber, S., Krawczak, M., Broring, C.D., Volzke, H., Schafmayer, C., Vodicka, P., Chang-Claude, J., Brenner, H.,

105
Burwinkel, B., Propping, P., Hampe, J., and Hemminki, K. (2010). Genome-wide association study for colorectal cancer identifies risk polymorphisms in German familial cases and implicates MAPK signalling pathways in disease susceptibility. Carcinogenesis 31, 1612-1619.
Latchford, A.R., Neale, K., Phillips, R.K., and Clark, S.K. (2011). Peutz-Jeghers syndrome: intriguing suggestion of gastrointestinal cancer prevention from surveillance. Dis Colon Rectum 54, 1547-1551.
Leblanc, P.M., Yeretssian, G., Rutherford, N., Doiron, K., Nadiri, A., Zhu, L., Green, D.R., Gruenheid, S., and Saleh, M. (2008). Caspase-12 modulates NOD signaling and regulates antimicrobial peptide production and mucosal immunity. Cell Host Microbe 3, 146-157.
Lecine, P., Esmiol, S., Metais, J.Y., Nicoletti, C., Nourry, C., Mcdonald, C., Nunez, G., Hugot, J.P., Borg, J.P., and Ollendorff, V. (2007). The NOD2-RICK complex signals from the plasma membrane. J Biol Chem 282, 15197-15207.
Lee, W.S., Baek, J.H., You, D.H., and Nam, M.J. (2013). Prognostic value of circulating cytokines for stage III colon cancer. J Surg Res 182, 49-54.
Li, D.M., and Sun, H. (1997). TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res 57, 2124-2129.
Li, J., Yen, C., Liaw, D., Podsypanina, K., Bose, S., Wang, S.I., Puc, J., Miliaresis, C., Rodgers, L., Mccombie, R., Bigner, S.H., Giovanella, B.C., Ittmann, M., Tycko, B., Hibshoosh, H., Wigler, M.H., and Parsons, R. (1997). PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 275, 1943-1947.
Li, M.O., Wan, Y.Y., and Flavell, R.A. (2007). T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579-591.
Lieberman, D.A., Rex, D.K., Winawer, S.J., Giardiello, F.M., Johnson, D.A., and Levin, T.R. (2012). Guidelines for colonoscopy surveillance after screening and polypectomy: a consensus update by the US Multi-Society Task Force on Colorectal Cancer. Gastroenterology 143, 844-857.
Ligtenberg, M.J., Kuiper, R.P., Chan, T.L., Goossens, M., Hebeda, K.M., Voorendt, M., Lee, T.Y., Bodmer, D., Hoenselaar, E., Hendriks-Cornelissen, S.J., Tsui, W.Y., Kong, C.K., Brunner, H.G., Van Kessel, A.G., Yuen, S.T., Van Krieken, J.H., Leung, S.Y., and Hoogerbrugge, N. (2009). Heritable somatic methylation and inactivation of MSH2 in families with Lynch syndrome due to deletion of the 3' exons of TACSTD1. Nat Genet 41, 112-117.
Lin-Marq, N., Borel, C., and Antonarakis, S.E. (2005). Peutz-Jeghers LKB1 mutants fail to activate GSK-3beta, preventing it from inhibiting Wnt signaling. Mol Genet Genomics 273, 184-196.
Lin, E.Y., Li, J.F., Gnatovskiy, L., Deng, Y., Zhu, L., Grzesik, D.A., Qian, H., Xue, X.N., and Pollard, J.W. (2006). Macrophages regulate the angiogenic switch in a mouse model of breast cancer. Cancer Res 66, 11238-11246.
Lin, J.T., Wang, J.Y., Chen, M.K., Chen, H.C., Chang, T.H., Su, B.W., and Chang, P.J. (2013). Colon cancer mesenchymal stem cells modulate the tumorigenicity of colon cancer through interleukin 6. Exp Cell Res 319, 2216-2229.
Lipinski, S., Grabe, N., Jacobs, G., Billmann-Born, S., Till, A., Hasler, R., Aden, K., Paulsen, M., Arlt, A., Kraemer, L., Hagemann, N., Erdmann, K.S., Schreiber, S., and Rosenstiel, P. (2012). RNAi screening identifies mediators of NOD2 signaling: implications for spatial specificity of MDP recognition. Proc Natl Acad Sci U S A 109, 21426-21431.
Liu, C., Li, Y., Semenov, M., Han, C., Baeg, G.H., Tan, Y., Zhang, Z., Lin, X., and He, X. (2002). Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108, 837-847.
Liu, Q., Hao, C., Su, P., and Shi, J. (2009). Down-regulation of HLA class I antigen-processing machinery components in esophageal squamous cell carcinomas: association with disease progression. Scand J Gastroenterol 44, 960-969.

106
Liu, Q.X., Wang, X.F., Ikeo, K., Hirose, S., Gehring, W.J., and Gojobori, T. (2014). Evolutionarily conserved transcription factor Apontic controls the G1/S progression by inducing cyclin E during eye development. Proc Natl Acad Sci U S A.
Luebeck, E.G., and Moolgavkar, S.H. (2002). Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci U S A 99, 15095-15100.
Luheshi, N., Davies, G., Poon, E., Wiggins, K., Mccourt, M., and Legg, J. (2014). Th1 cytokines are more effective than Th2 cytokines at licensing anti-tumour functions in CD40-activated human macrophages in vitro. Eur J Immunol 44, 162-172.
Mabbott, N.A., Donaldson, D.S., Ohno, H., Williams, I.R., and Mahajan, A. (2013). Microfold (M) cells: important immunosurveillance posts in the intestinal epithelium. Mucosal Immunol 6, 666-677.
Magner, W.J., Kazim, A.L., Stewart, C., Romano, M.A., Catalano, G., Grande, C., Keiser, N., Santaniello, F., and Tomasi, T.B. (2000). Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol 165, 7017-7024.
Mantovani, A., and Locati, M. (2013). Tumor-associated macrophages as a paradigm of macrophage plasticity, diversity, and polarization: lessons and open questions. Arterioscler Thromb Vasc Biol 33, 1478-1483.
Mao, Q., Zhang, Y., Fu, X., Xue, J., Guo, W., Meng, M., Zhou, Z., Mo, X., and Lu, Y. (2013). A tumor hypoxic niche protects human colon cancer stem cells from chemotherapy. J Cancer Res Clin Oncol 139, 211-222.
Markowitz, S., Wang, J., Myeroff, L., Parsons, R., Sun, L., Lutterbaugh, J., Fan, R.S., Zborowska, E., Kinzler, K.W., Vogelstein, B., and Et Al. (1995). Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 268, 1336-1338.
Matise, L.A., Palmer, T.D., Ashby, W.J., Nashabi, A., Chytil, A., Aakre, M., Pickup, M.W., Gorska, A.E., Zijlstra, A., and Moses, H.L. (2012). Lack of transforming growth factor-beta signaling promotes collective cancer cell invasion through tumor-stromal crosstalk. Breast Cancer Res 14, R98.
Mendoza, M.C., Er, E.E., and Blenis, J. (2011). The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 36, 320-328.
Miles, F.L., and Sikes, R.A. (2014). Insidious changes in stromal matrix fuel cancer progression. Mol Cancer Res 12, 297-312.
Mink, S.R., Vashistha, S., Zhang, W., Hodge, A., Agus, D.B., and Jain, A. (2010). Cancer-associated fibroblasts derived from EGFR-TKI-resistant tumors reverse EGFR pathway inhibition by EGFR-TKIs. Mol Cancer Res 8, 809-820.
Mita, M.M., Sargsyan, L., Mita, A.C., and Spear, M. (2013). Vascular-disrupting agents in oncology. Expert Opin Investig Drugs 22, 317-328.
Mittal, D., Gubin, M.M., Schreiber, R.D., and Smyth, M.J. (2014). New insights into cancer immunoediting and its three component phases--elimination, equilibrium and escape. Curr Opin Immunol 27, 16-25.
Miyaki, M., Iijima, T., Konishi, M., Sakai, K., Ishii, A., Yasuno, M., Hishima, T., Koike, M., Shitara, N., Iwama, T., Utsunomiya, J., Kuroki, T., and Mori, T. (1999). Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene 18, 3098-3103.
Mombaerts, P., Iacomini, J., Johnson, R.S., Herrup, K., Tonegawa, S., and Papaioannou, V.E. (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869-877.
Monticelli, L.A., Sonnenberg, G.F., Abt, M.C., Alenghat, T., Ziegler, C.G., Doering, T.A., Angelosanto, J.M., Laidlaw, B.J., Yang, C.Y., Sathaliyawala, T., Kubota, M., Turner, D., Diamond, J.M., Goldrath, A.W., Farber, D.L., Collman, R.G., Wherry, E.J., and Artis, D. (2011). Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12, 1045-1054.

107
Moon, B.S., Jeong, W.J., Park, J., Kim, T.I., Min Do, S., and Choi, K.Y. (2014). Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/beta-catenin signaling. J Natl Cancer Inst 106, djt373.
Morin, P.J., Sparks, A.B., Korinek, V., Barker, N., Clevers, H., Vogelstein, B., and Kinzler, K.W. (1997). Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275, 1787-1790.
Morton, D.G., Roos, J.M., and Kemphues, K.J. (1992). par-4, a gene required for cytoplasmic localization and determination of specific cell types in Caenorhabditis elegans embryogenesis. Genetics 130, 771-790.
Mouradov, D., Domingo, E., Gibbs, P., Jorissen, R.N., Li, S., Soo, P.Y., Lipton, L., Desai, J., Danielsen, H.E., Oukrif, D., Novelli, M., Yau, C., Holmes, C.C., Jones, I.T., Mclaughlin, S., Molloy, P., Hawkins, N.J., Ward, R., Midgely, R., Kerr, D., Tomlinson, I.P., and Sieber, O.M. (2013). Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol 108, 1785-1793.
Mumert, M., Dubuc, A., Wu, X., Northcott, P.A., Chin, S.S., Pedone, C.A., Taylor, M.D., and Fults, D.W. (2012). Functional genomics identifies drivers of medulloblastoma dissemination. Cancer Res 72, 4944-4953.
Musgrove, E.A., Caldon, C.E., Barraclough, J., Stone, A., and Sutherland, R.L. (2011). Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11, 558-572.
Nakagawa, T., Zhu, H., Morishima, N., Li, E., Xu, J., Yankner, B.A., and Yuan, J. (2000). Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature 403, 98-103.
Narunsky, L., Oren, R., Bochner, F., and Neeman, M. (2014). Imaging aspects of the tumor stroma with therapeutic implications. Pharmacol Ther 141, 192-208.
Nathan, P., Zweifel, M., Padhani, A.R., Koh, D.M., Ng, M., Collins, D.J., Harris, A., Carden, C., Smythe, J., Fisher, N., Taylor, N.J., Stirling, J.J., Lu, S.P., Leach, M.O., Rustin, G.J., and Judson, I. (2012). Phase I trial of combretastatin A4 phosphate (CA4P) in combination with bevacizumab in patients with advanced cancer. Clin Cancer Res 18, 3428-3439.
Nembrini, C., Kisielow, J., Shamshiev, A.T., Tortola, L., Coyle, A.J., Kopf, M., and Marsland, B.J. (2009). The kinase activity of Rip2 determines its stability and consequently Nod1- and Nod2-mediated immune responses. J Biol Chem 284, 19183-19188.
Ness, J.M., Harvey, C.A., Strasser, A., Bouillet, P., Klocke, B.J., and Roth, K.A. (2006). Selective involvement of BH3-only Bcl-2 family members Bim and Bad in neonatal hypoxia-ischemia. Brain Res 1099, 150-159.
Newton, K., Sun, X., and Dixit, V.M. (2004). Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol 24, 1464-1469.
Nilsson, S., Moller, C., Jirstrom, K., Lee, A., Busch, S., Lamb, R., and Landberg, G. (2012). Downregulation of miR-92a is associated with aggressive breast cancer features and increased tumour macrophage infiltration. PLoS One 7, e36051.
Nosho, K., Irahara, N., Shima, K., Kure, S., Kirkner, G.J., Schernhammer, E.S., Hazra, A., Hunter, D.J., Quackenbush, J., Spiegelman, D., Giovannucci, E.L., Fuchs, C.S., and Ogino, S. (2008). Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS One 3, e3698.
Nozawa, H., Chiu, C., and Hanahan, D. (2006). Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc Natl Acad Sci U S A 103, 12493-12498.

108
Nucci, M.R., Robinson, C.R., Longo, P., Campbell, P., and Hamilton, S.R. (1997). Phenotypic and genotypic characteristics of aberrant crypt foci in human colorectal mucosa. Hum Pathol 28, 1396-1407.
Obermajer, N., Muthuswamy, R., Lesnock, J., Edwards, R.P., and Kalinski, P. (2011). Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 118, 5498-5505.
Oliveira, C., Velho, S., Moutinho, C., Ferreira, A., Preto, A., Domingo, E., Capelinha, A.F., Duval, A., Hamelin, R., Machado, J.C., Schwartz, S., Jr., Carneiro, F., and Seruca, R. (2007). KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene 26, 158-163.
Ong, S.M., Tan, Y.C., Beretta, O., Jiang, D., Yeap, W.H., Tai, J.J., Wong, W.C., Yang, H., Schwarz, H., Lim, K.H., Koh, P.K., Ling, K.L., and Wong, S.C. (2012). Macrophages in human colorectal cancer are pro-inflammatory and prime T cells towards an anti-tumour type-1 inflammatory response. Eur J Immunol 42, 89-100.
Paez-Ribes, M., Allen, E., Hudock, J., Takeda, T., Okuyama, H., Vinals, F., Inoue, M., Bergers, G., Hanahan, D., and Casanovas, O. (2009). Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220-231.
Paget, G. (1889). Remarks on a Case of Alternate Partial Anaesthesia. Br Med J 1, 1-3. Pan, P.Y., Ma, G., Weber, K.J., Ozao-Choy, J., Wang, G., Yin, B., Divino, C.M., and Chen, S.H. (2010).
Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res 70, 99-108.
Pande, M., Lynch, P.M., Hopper, J.L., Jenkins, M.A., Gallinger, S., Haile, R.W., Lemarchand, L., Lindor, N.M., Campbell, P.T., Newcomb, P.A., Potter, J.D., Baron, J.A., Frazier, M.L., and Amos, C.I. (2010). Smoking and colorectal cancer in Lynch syndrome: results from the Colon Cancer Family Registry and the University of Texas M.D. Anderson Cancer Center. Clin Cancer Res 16, 1331-1339.
Park, J.H., Kim, Y.G., Shaw, M., Kanneganti, T.D., Fujimoto, Y., Fukase, K., Inohara, N., and Nunez, G. (2007). Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J Immunol 179, 514-521.
Parry, S., Win, A.K., Parry, B., Macrae, F.A., Gurrin, L.C., Church, J.M., Baron, J.A., Giles, G.G., Leggett, B.A., Winship, I., Lipton, L., Young, G.P., Young, J.P., Lodge, C.J., Southey, M.C., Newcomb, P.A., Le Marchand, L., Haile, R.W., Lindor, N.M., Gallinger, S., Hopper, J.L., and Jenkins, M.A. (2011). Metachronous colorectal cancer risk for mismatch repair gene mutation carriers: the advantage of more extensive colon surgery. Gut 60, 950-957.
Pauleau, A.L., and Murray, P.J. (2003). Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol Cell Biol 23, 7531-7539.
Pedemonte, S., Sciallero, S., Gismondi, V., Stagnaro, P., Biticchi, R., Haeouaine, A., Bonelli, L., Nicolo, G., Groden, J., Bruzzi, P., Aste, H., and Varesco, L. (1998). Novel germline APC variants in patients with multiple adenomas. Genes Chromosomes Cancer 22, 257-267.
Peng, Y., Wang, L., and Gu, J. (2013). Elevated preoperative carcinoembryonic antigen (CEA) and Ki67 is predictor of decreased survival in IIA stage colon cancer. World J Surg 37, 208-213.
Peters, J.M., Mckay, R.M., Mckay, J.P., and Graff, J.M. (1999). Casein kinase I transduces Wnt signals. Nature 401, 345-350.
Peters, U., Hutter, C.M., Hsu, L., Schumacher, F.R., Conti, D.V., Carlson, C.S., Edlund, C.K., Haile, R.W., Gallinger, S., Zanke, B.W., Lemire, M., Rangrej, J., Vijayaraghavan, R., Chan, A.T., Hazra, A., Hunter, D.J., Ma, J., Fuchs, C.S., Giovannucci, E.L., Kraft, P., Liu, Y., Chen, L., Jiao, S., Makar, K.W., Taverna, D., Gruber, S.B., Rennert, G., Moreno, V., Ulrich, C.M., Woods, M.O., Green, R.C., Parfrey, P.S., Prentice, R.L., Kooperberg, C., Jackson, R.D., Lacroix, A.Z., Caan, B.J., Hayes, R.B., Berndt, S.I., Chanock, S.J., Schoen, R.E., Chang-Claude, J., Hoffmeister, M., Brenner, H., Frank, B., Bezieau, S., Kury, S., Slattery, M.L., Hopper, J.L., Jenkins, M.A., Le Marchand, L., Lindor, N.M.,

109
Newcomb, P.A., Seminara, D., Hudson, T.J., Duggan, D.J., Potter, J.D., and Casey, G. (2012). Meta-analysis of new genome-wide association studies of colorectal cancer risk. Hum Genet 131, 217-234.
Peters, U., Jiao, S., Schumacher, F.R., Hutter, C.M., Aragaki, A.K., Baron, J.A., Berndt, S.I., Bezieau, S., Brenner, H., Butterbach, K., Caan, B.J., Campbell, P.T., Carlson, C.S., Casey, G., Chan, A.T., Chang-Claude, J., Chanock, S.J., Chen, L.S., Coetzee, G.A., Coetzee, S.G., Conti, D.V., Curtis, K.R., Duggan, D., Edwards, T., Fuchs, C.S., Gallinger, S., Giovannucci, E.L., Gogarten, S.M., Gruber, S.B., Haile, R.W., Harrison, T.A., Hayes, R.B., Henderson, B.E., Hoffmeister, M., Hopper, J.L., Hudson, T.J., Hunter, D.J., Jackson, R.D., Jee, S.H., Jenkins, M.A., Jia, W.H., Kolonel, L.N., Kooperberg, C., Kury, S., Lacroix, A.Z., Laurie, C.C., Laurie, C.A., Le Marchand, L., Lemire, M., Levine, D., Lindor, N.M., Liu, Y., Ma, J., Makar, K.W., Matsuo, K., Newcomb, P.A., Potter, J.D., Prentice, R.L., Qu, C., Rohan, T., Rosse, S.A., Schoen, R.E., Seminara, D., Shrubsole, M., Shu, X.O., Slattery, M.L., Taverna, D., Thibodeau, S.N., Ulrich, C.M., White, E., Xiang, Y., Zanke, B.W., Zeng, Y.X., Zhang, B., Zheng, W., and Hsu, L. (2013). Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology 144, 799-807 e724.
Peterson, L.W., and Artis, D. (2014). Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol 14, 141-153.
Petitjean, A., Mathe, E., Kato, S., Ishioka, C., Tavtigian, S.V., Hainaut, P., and Olivier, M. (2007). Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat 28, 622-629.
Pull, S.L., Doherty, J.M., Mills, J.C., Gordon, J.I., and Stappenbeck, T.S. (2005). Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A 102, 99-104.
Pylayeva-Gupta, Y., Lee, K.E., Hajdu, C.H., Miller, G., and Bar-Sagi, D. (2012). Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 21, 836-847.
Qiao, L., and Wong, B.C. (2009). Role of Notch signaling in colorectal cancer. Carcinogenesis 30, 1979-1986.
Quehenberger, F., Vasen, H.F., and Van Houwelingen, H.C. (2005). Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 42, 491-496.
Qureshi, O.S., Zheng, Y., Nakamura, K., Attridge, K., Manzotti, C., Schmidt, E.M., Baker, J., Jeffery, L.E., Kaur, S., Briggs, Z., Hou, T.Z., Futter, C.E., Anderson, G., Walker, L.S., and Sansom, D.M. (2011). Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332, 600-603.
Quwailid, M.M., Hugill, A., Dear, N., Vizor, L., Wells, S., Horner, E., Fuller, S., Weedon, J., Mcmath, H., Woodman, P., Edwards, D., Campbell, D., Rodger, S., Carey, J., Roberts, A., Glenister, P., Lalanne, Z., Parkinson, N., Coghill, E.L., Mckeone, R., Cox, S., Willan, J., Greenfield, A., Keays, D., Brady, S., Spurr, N., Gray, I., Hunter, J., Brown, S.D., and Cox, R.D. (2004). A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm Genome 15, 585-591.
Rattigan, Y.I., Patel, B.B., Ackerstaff, E., Sukenick, G., Koutcher, J.A., Glod, J.W., and Banerjee, D. (2012). Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp Cell Res 318, 326-335.
Renz, H., Brandtzaeg, P., and Hornef, M. (2012). The impact of perinatal immune development on mucosal homeostasis and chronic inflammation. Nat Rev Immunol 12, 9-23.
Reynolds, A., Wharton, N., Parris, A., Mitchell, E., Sobolewski, A., Kam, C., Bigwood, L., El Hadi, A., Munsterberg, A., Lewis, M., Speakman, C., Stebbings, W., Wharton, R., Sargen, K., Tighe, R., Jamieson, C., Hernon, J., Kapur, S., Oue, N., Yasui, W., and Williams, M.R. (2014). Canonical Wnt

110
signals combined with suppressed TGFbeta/BMP pathways promote renewal of the native human colonic epithelium. Gut 63, 610-621.
Ribas, A., Kefford, R., Marshall, M.A., Punt, C.J., Haanen, J.B., Marmol, M., Garbe, C., Gogas, H., Schachter, J., Linette, G., Lorigan, P., Kendra, K.L., Maio, M., Trefzer, U., Smylie, M., Mcarthur, G.A., Dreno, B., Nathan, P.D., Mackiewicz, J., Kirkwood, J.M., Gomez-Navarro, J., Huang, B., Pavlov, D., and Hauschild, A. (2013). Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol 31, 616-622.
Rinchik, E.M., Carpenter, D.A., and Selby, P.B. (1990). A strategy for fine-structure functional analysis of a 6- to 11-centimorgan region of mouse chromosome 7 by high-efficiency mutagenesis. Proc Natl Acad Sci U S A 87, 896-900.
Robert, C., Thomas, L., Bondarenko, I., O'day, S., M, D.J., Garbe, C., Lebbe, C., Baurain, J.F., Testori, A., Grob, J.J., Davidson, N., Richards, J., Maio, M., Hauschild, A., Miller, W.H., Jr., Gascon, P., Lotem, M., Harmankaya, K., Ibrahim, R., Francis, S., Chen, T.T., Humphrey, R., Hoos, A., and Wolchok, J.D. (2011). Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364, 2517-2526.
Rollmann, S.M., Yamamoto, A., Goossens, T., Zwarts, L., Callaerts-Vegh, Z., Callaerts, P., Norga, K., Mackay, T.F., and Anholt, R.R. (2007). The early developmental gene Semaphorin 5c contributes to olfactory behavior in adult Drosophila. Genetics 176, 947-956.
Ronnov-Jessen, L., Petersen, O.W., and Bissell, M.J. (1996). Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev 76, 69-125.
Rosenberg, D.W., Giardina, C., and Tanaka, T. (2009). Mouse models for the study of colon carcinogenesis. Carcinogenesis 30, 183-196.
Rost, M., Doring, T., and Prange, R. (2008). gamma2-Adaptin, a ubiquitin-interacting adaptor, is a substrate to coupled ubiquitination by the ubiquitin ligase Nedd4 and functions in the endosomal pathway. J Biol Chem 283, 32119-32130.
Roth, A.D., Tejpar, S., Delorenzi, M., Yan, P., Fiocca, R., Klingbiel, D., Dietrich, D., Biesmans, B., Bodoky, G., Barone, C., Aranda, E., Nordlinger, B., Cisar, L., Labianca, R., Cunningham, D., Van Cutsem, E., and Bosman, F. (2010). Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 28, 466-474.
Rothwell, P.M., Fowkes, F.G., Belch, J.F., Ogawa, H., Warlow, C.P., and Meade, T.W. (2011). Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet 377, 31-41.
Said-Sadier, N., and Ojcius, D.M. (2012). Alarmins, inflammasomes and immunity. Biomed J 35, 437-449. Sakanaka, C., Leong, P., Xu, L., Harrison, S.D., and Williams, L.T. (1999). Casein kinase iepsilon in the wnt
pathway: regulation of beta-catenin function. Proc Natl Acad Sci U S A 96, 12548-12552. Saleh, M., Mathison, J.C., Wolinski, M.K., Bensinger, S.J., Fitzgerald, P., Droin, N., Ulevitch, R.J., Green,
D.R., and Nicholson, D.W. (2006). Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature 440, 1064-1068.
Saltz, L.B., Clarke, S., Diaz-Rubio, E., Scheithauer, W., Figer, A., Wong, R., Koski, S., Lichinitser, M., Yang, T.S., Rivera, F., Couture, F., Sirzen, F., and Cassidy, J. (2008). Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26, 2013-2019.
Samowitz, W.S., Albertsen, H., Sweeney, C., Herrick, J., Caan, B.J., Anderson, K.E., Wolff, R.K., and Slattery, M.L. (2006). Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst 98, 1731-1738.

111
Sato, T., Van Es, J.H., Snippert, H.J., Stange, D.E., Vries, R.G., Van Den Born, M., Barker, N., Shroyer, N.F., Van De Wetering, M., and Clevers, H. (2011). Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 469, 415-418.
Sceneay, J., Chow, M.T., Chen, A., Halse, H.M., Wong, C.S., Andrews, D.M., Sloan, E.K., Parker, B.S., Bowtell, D.D., Smyth, M.J., and Moller, A. (2012). Primary tumor hypoxia recruits CD11b+/Ly6Cmed/Ly6G+ immune suppressor cells and compromises NK cell cytotoxicity in the premetastatic niche. Cancer Res 72, 3906-3911.
Sceneay, J., Smyth, M.J., and Moller, A. (2013). The pre-metastatic niche: finding common ground. Cancer Metastasis Rev 32, 449-464.
Schneider, H., Valk, E., Da Rocha Dias, S., Wei, B., and Rudd, C.E. (2005). CTLA-4 up-regulation of lymphocyte function-associated antigen 1 adhesion and clustering as an alternate basis for coreceptor function. Proc Natl Acad Sci U S A 102, 12861-12866.
Schreiber, R.D., Old, L.J., and Smyth, M.J. (2011). Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science 331, 1565-1570.
Schwarz-Romond, T., Fiedler, M., Shibata, N., Butler, P.J., Kikuchi, A., Higuchi, Y., and Bienz, M. (2007). The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat Struct Mol Biol 14, 484-492.
Serafini, P., Mgebroff, S., Noonan, K., and Borrello, I. (2008). Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res 68, 5439-5449.
Seshagiri, S., Stawiski, E.W., Durinck, S., Modrusan, Z., Storm, E.E., Conboy, C.B., Chaudhuri, S., Guan, Y., Janakiraman, V., Jaiswal, B.S., Guillory, J., Ha, C., Dijkgraaf, G.J., Stinson, J., Gnad, F., Huntley, M.A., Degenhardt, J.D., Haverty, P.M., Bourgon, R., Wang, W., Koeppen, H., Gentleman, R., Starr, T.K., Zhang, Z., Largaespada, D.A., Wu, T.D., and De Sauvage, F.J. (2012). Recurrent R-spondin fusions in colon cancer. Nature 488, 660-664.
Shankaran, V., Ikeda, H., Bruce, A.T., White, J.M., Swanson, P.E., Old, L.J., and Schreiber, R.D. (2001). IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107-1111.
Shih, T., and Lindley, C. (2006). Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther 28, 1779-1802.
Shojaei, F., Wu, X., Qu, X., Kowanetz, M., Yu, L., Tan, M., Meng, Y.G., and Ferrara, N. (2009). G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proc Natl Acad Sci U S A 106, 6742-6747.
Shojaei, F., Wu, X., Zhong, C., Yu, L., Liang, X.H., Yao, J., Blanchard, D., Bais, C., Peale, F.V., Van Bruggen, N., Ho, C., Ross, J., Tan, M., Carano, R.A., Meng, Y.G., and Ferrara, N. (2007). Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450, 825-831.
Shurin, G.V., Ma, Y., and Shurin, M.R. (2013). Immunosuppressive mechanisms of regulatory dendritic cells in cancer. Cancer Microenviron 6, 159-167.
Simms, N.A., Rajput, A., Sharratt, E.A., Ongchin, M., Teggart, C.A., Wang, J., and Brattain, M.G. (2012). Transforming growth factor-beta suppresses metastasis in a subset of human colon carcinoma cells. BMC Cancer 12, 221.
Singel, S.M., Batten, K., Cornelius, C., Jia, G., Fasciani, G., Barron, S.L., Wright, W.E., and Shay, J.W. (2014). Receptor-interacting protein kinase 2 promotes triple-negative breast cancer cell migration and invasion via activation of nuclear factor-kappaB and c-Jun N-terminal kinase pathways. Breast Cancer Res 16, R28.
Slattery, M.L., John, E.M., Torres-Mejia, G., Lundgreen, A., Lewinger, J.P., Stern, M.C., Hines, L., Baumgartner, K.B., Giuliano, A.R., and Wolff, R.K. (2014). Angiogenesis genes, dietary oxidative balance and breast cancer risk and progression: the Breast Cancer Health Disparities Study. Int J Cancer 134, 629-644.

112
Smith, J.J., Deane, N.G., Wu, F., Merchant, N.B., Zhang, B., Jiang, A., Lu, P., Johnson, J.C., Schmidt, C., Bailey, C.E., Eschrich, S., Kis, C., Levy, S., Washington, M.K., Heslin, M.J., Coffey, R.J., Yeatman, T.J., Shyr, Y., and Beauchamp, R.D. (2010). Experimentally derived metastasis gene expression profile predicts recurrence and death in patients with colon cancer. Gastroenterology 138, 958-968.
Smythies, L.E., Sellers, M., Clements, R.H., Mosteller-Barnum, M., Meng, G., Benjamin, W.H., Orenstein, J.M., and Smith, P.D. (2005). Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115, 66-75.
Soravia, C., Berk, T., Madlensky, L., Mitri, A., Cheng, H., Gallinger, S., Cohen, Z., and Bapat, B. (1998). Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 62, 1290-1301.
Spirio, L., Olschwang, S., Groden, J., Robertson, M., Samowitz, W., Joslyn, G., Gelbert, L., Thliveris, A., Carlson, M., Otterud, B., and Et Al. (1993). Alleles of the APC gene: an attenuated form of familial polyposis. Cell 75, 951-957.
Spranger, S., Spaapen, R.M., Zha, Y., Williams, J., Meng, Y., Ha, T.T., and Gajewski, T.F. (2013). Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 5, 200ra116.
Stacey, S.N., Sulem, P., Jonasdottir, A., Masson, G., Gudmundsson, J., Gudbjartsson, D.F., Magnusson, O.T., Gudjonsson, S.A., Sigurgeirsson, B., Thorisdottir, K., Ragnarsson, R., Benediktsdottir, K.R., Nexo, B.A., Tjonneland, A., Overvad, K., Rudnai, P., Gurzau, E., Koppova, K., Hemminki, K., Corredera, C., Fuentelsaz, V., Grasa, P., Navarrete, S., Fuertes, F., Garcia-Prats, M.D., Sanambrosio, E., Panadero, A., De Juan, A., Garcia, A., Rivera, F., Planelles, D., Soriano, V., Requena, C., Aben, K.K., Van Rossum, M.M., Cremers, R.G., Van Oort, I.M., Van Spronsen, D.J., Schalken, J.A., Peters, W.H., Helfand, B.T., Donovan, J.L., Hamdy, F.C., Badescu, D., Codreanu, O., Jinga, M., Csiki, I.E., Constantinescu, V., Badea, P., Mates, I.N., Dinu, D.E., Constantin, A., Mates, D., Kristjansdottir, S., Agnarsson, B.A., Jonsson, E., Barkardottir, R.B., Einarsson, G.V., Sigurdsson, F., Moller, P.H., Stefansson, T., Valdimarsson, T., Johannsson, O.T., Sigurdsson, H., Jonsson, T., Jonasson, J.G., Tryggvadottir, L., Rice, T., Hansen, H.M., Xiao, Y., Lachance, D.H., Bp, O.N., Kosel, M.L., Decker, P.A., Thorleifsson, G., Johannsdottir, H., Helgadottir, H.T., Sigurdsson, A., Steinthorsdottir, V., Lindblom, A., Sandler, R.S., Keku, T.O., Banasik, K., Jorgensen, T., Witte, D.R., Hansen, T., Pedersen, O., Jinga, V., Neal, D.E., Catalona, W.J., Wrensch, M., Wiencke, J., Jenkins, R.B., Nagore, E., Vogel, U., Kiemeney, L.A., Kumar, R., Mayordomo, J.I., Olafsson, J.H., Kong, A., et al. (2011). A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat Genet 43, 1098-1103.
Steck, P.A., Pershouse, M.A., Jasser, S.A., Yung, W.K., Lin, H., Ligon, A.H., Langford, L.A., Baumgard, M.L., Hattier, T., Davis, T., Frye, C., Hu, R., Swedlund, B., Teng, D.H., and Tavtigian, S.V. (1997). Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15, 356-362.
Stegeman, I., De Wijkerslooth, T.R., Stoop, E.M., Van Leerdam, M.E., Dekker, E., Van Ballegooijen, M., Kuipers, E.J., Fockens, P., Kraaijenhagen, R.A., and Bossuyt, P.M. (2013). Colorectal cancer risk factors in the detection of advanced adenoma and colorectal cancer. Cancer Epidemiol 37, 278-283.
Sturgeon, D., Mccutcheon, T., Geiger, T.M., Muldoon, R.L., Herline, A.J., and Wise, P.E. (2013). Increasing Lynch syndrome identification through establishment of a hereditary colorectal cancer registry. Dis Colon Rectum 56, 308-314.
Swiderska, M., Choromanska, B., Dabrowska, E., Konarzewska-Duchnowska, E., Choromanska, K., Szczurko, G., Mysliwiec, P., Dadan, J., Ladny, J.R., and Zwierz, K. (2014). The diagnostics of colorectal cancer. Contemp Oncol (Pozn) 18, 1-6.

113
Takatsu, H., Sakurai, M., Shin, H.W., Murakami, K., and Nakayama, K. (1998). Identification and characterization of novel clathrin adaptor-related proteins. J Biol Chem 273, 24693-24700.
Tanabe, Y., Fujita, E., Hayashi, Y.K., Zhu, X., Lubbert, H., Mezaki, Y., Senoo, H., and Momoi, T. (2013). Synaptic adhesion molecules in Cadm family at the neuromuscular junction. Cell Biol Int 37, 731-736.
Tao, M., Scacheri, P.C., Marinis, J.M., Harhaj, E.W., Matesic, L.E., and Abbott, D.W. (2009). ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol 19, 1255-1263.
Tattoli, I., Travassos, L.H., Carneiro, L.A., Magalhaes, J.G., and Girardin, S.E. (2007). The Nodosome: Nod1 and Nod2 control bacterial infections and inflammation. Semin Immunopathol 29, 289-301.
Tenesa, A., Farrington, S.M., Prendergast, J.G., Porteous, M.E., Walker, M., Haq, N., Barnetson, R.A., Theodoratou, E., Cetnarskyj, R., Cartwright, N., Semple, C., Clark, A.J., Reid, F.J., Smith, L.A., Kavoussanakis, K., Koessler, T., Pharoah, P.D., Buch, S., Schafmayer, C., Tepel, J., Schreiber, S., Volzke, H., Schmidt, C.O., Hampe, J., Chang-Claude, J., Hoffmeister, M., Brenner, H., Wilkening, S., Canzian, F., Capella, G., Moreno, V., Deary, I.J., Starr, J.M., Tomlinson, I.P., Kemp, Z., Howarth, K., Carvajal-Carmona, L., Webb, E., Broderick, P., Vijayakrishnan, J., Houlston, R.S., Rennert, G., Ballinger, D., Rozek, L., Gruber, S.B., Matsuda, K., Kidokoro, T., Nakamura, Y., Zanke, B.W., Greenwood, C.M., Rangrej, J., Kustra, R., Montpetit, A., Hudson, T.J., Gallinger, S., Campbell, H., and Dunlop, M.G. (2008). Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet 40, 631-637.
Teng, M.W., Vesely, M.D., Duret, H., Mclaughlin, N., Towne, J.E., Schreiber, R.D., and Smyth, M.J. (2012). Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res 72, 3987-3996.
Thompson-Fawcett, M.W., Marcus, V.A., Redston, M., Cohen, Z., and Mcleod, R.S. (2001). Adenomatous polyps develop commonly in the ileal pouch of patients with familial adenomatous polyposis. Dis Colon Rectum 44, 347-353.
Tiainen, M., Vaahtomeri, K., Ylikorkala, A., and Makela, T.P. (2002). Growth arrest by the LKB1 tumor suppressor: induction of p21(WAF1/CIP1). Hum Mol Genet 11, 1497-1504.
Tiainen, M., Ylikorkala, A., and Makela, T.P. (1999). Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci U S A 96, 9248-9251.
Tlsty, T.D., and Hein, P.W. (2001). Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev 11, 54-59.
Tomlinson, I.P., Carvajal-Carmona, L.G., Dobbins, S.E., Tenesa, A., Jones, A.M., Howarth, K., Palles, C., Broderick, P., Jaeger, E.E., Farrington, S., Lewis, A., Prendergast, J.G., Pittman, A.M., Theodoratou, E., Olver, B., Walker, M., Penegar, S., Barclay, E., Whiffin, N., Martin, L., Ballereau, S., Lloyd, A., Gorman, M., Lubbe, S., Howie, B., Marchini, J., Ruiz-Ponte, C., Fernandez-Rozadilla, C., Castells, A., Carracedo, A., Castellvi-Bel, S., Duggan, D., Conti, D., Cazier, J.B., Campbell, H., Sieber, O., Lipton, L., Gibbs, P., Martin, N.G., Montgomery, G.W., Young, J., Baird, P.N., Gallinger, S., Newcomb, P., Hopper, J., Jenkins, M.A., Aaltonen, L.A., Kerr, D.J., Cheadle, J., Pharoah, P., Casey, G., Houlston, R.S., and Dunlop, M.G. (2011). Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet 7, e1002105.
Tomlinson, I.P., Webb, E., Carvajal-Carmona, L., Broderick, P., Howarth, K., Pittman, A.M., Spain, S., Lubbe, S., Walther, A., Sullivan, K., Jaeger, E., Fielding, S., Rowan, A., Vijayakrishnan, J., Domingo, E., Chandler, I., Kemp, Z., Qureshi, M., Farrington, S.M., Tenesa, A., Prendergast, J.G., Barnetson, R.A., Penegar, S., Barclay, E., Wood, W., Martin, L., Gorman, M., Thomas, H., Peto, J., Bishop, D.T., Gray, R., Maher, E.R., Lucassen, A., Kerr, D., Evans, D.G., Schafmayer, C., Buch, S., Volzke, H., Hampe, J., Schreiber, S., John, U., Koessler, T., Pharoah, P., Van Wezel, T., Morreau, H.,

114
Wijnen, J.T., Hopper, J.L., Southey, M.C., Giles, G.G., Severi, G., Castellvi-Bel, S., Ruiz-Ponte, C., Carracedo, A., Castells, A., Forsti, A., Hemminki, K., Vodicka, P., Naccarati, A., Lipton, L., Ho, J.W., Cheng, K.K., Sham, P.C., Luk, J., Agundez, J.A., Ladero, J.M., De La Hoya, M., Caldes, T., Niittymaki, I., Tuupanen, S., Karhu, A., Aaltonen, L., Cazier, J.B., Campbell, H., Dunlop, M.G., and Houlston, R.S. (2008). A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet 40, 623-630.
Topalian, S.L., Hodi, F.S., Brahmer, J.R., Gettinger, S.N., Smith, D.C., Mcdermott, D.F., Powderly, J.D., Carvajal, R.D., Sosman, J.A., Atkins, M.B., Leming, P.D., Spigel, D.R., Antonia, S.J., Horn, L., Drake, C.G., Pardoll, D.M., Chen, L., Sharfman, W.H., Anders, R.A., Taube, J.M., Mcmiller, T.L., Xu, H., Korman, A.J., Jure-Kunkel, M., Agrawal, S., Mcdonald, D., Kollia, G.D., Gupta, A., Wigginton, J.M., and Sznol, M. (2012). Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366, 2443-2454.
Toriola, A.T., Cheng, T.Y., Neuhouser, M.L., Wener, M.H., Zheng, Y., Brown, E., Miller, J.W., Song, X., Beresford, S.A., Gunter, M.J., Caudill, M.A., and Ulrich, C.M. (2013). Biomarkers of inflammation are associated with colorectal cancer risk in women but are not suitable as early detection markers. Int J Cancer 132, 2648-2658.
Toyota, M., Ahuja, N., Ohe-Toyota, M., Herman, J.G., Baylin, S.B., and Issa, J.P. (1999). CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 96, 8681-8686.
Tsikitis, V.L., Larson, D.W., Huebner, M., Lohse, C.M., and Thompson, P.A. (2014). Predictors of recurrence free survival for patients with stage II and III colon cancer. BMC Cancer 14, 336.
Udagawa, T. (2008). Tumor dormancy of primary and secondary cancers. APMIS 116, 615-628. Ugurel, S., Uhlig, D., Pfohler, C., Tilgen, W., Schadendorf, D., and Reinhold, U. (2004). Down-regulation of
HLA class II and costimulatory CD86/B7-2 on circulating monocytes from melanoma patients. Cancer Immunol Immunother 53, 551-559.
Van Der Flier, L.G., and Clevers, H. (2009). Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol 71, 241-260.
Van Hattem, W.A., Brosens, L.A., De Leng, W.W., Morsink, F.H., Lens, S., Carvalho, R., Giardiello, F.M., and Offerhaus, G.J. (2008). Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut 57, 623-627.
Vandooren, J., Van Den Steen, P.E., and Opdenakker, G. (2013). Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol 48, 222-272.
Vasen, H.F., Blanco, I., Aktan-Collan, K., Gopie, J.P., Alonso, A., Aretz, S., Bernstein, I., Bertario, L., Burn, J., Capella, G., Colas, C., Engel, C., Frayling, I.M., Genuardi, M., Heinimann, K., Hes, F.J., Hodgson, S.V., Karagiannis, J.A., Lalloo, F., Lindblom, A., Mecklin, J.P., Moller, P., Myrhoj, T., Nagengast, F.M., Parc, Y., Ponz De Leon, M., Renkonen-Sinisalo, L., Sampson, J.R., Stormorken, A., Sijmons, R.H., Tejpar, S., Thomas, H.J., Rahner, N., Wijnen, J.T., Jarvinen, H.J., and Moslein, G. (2013). Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 62, 812-823.
Vermeulen, L., De Sousa, E.M.F., Van Der Heijden, M., Cameron, K., De Jong, J.H., Borovski, T., Tuynman, J.B., Todaro, M., Merz, C., Rodermond, H., Sprick, M.R., Kemper, K., Richel, D.J., Stassi, G., and Medema, J.P. (2010). Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol 12, 468-476.
Villeneuve, P.J., and Sundaresan, R.S. (2009). Surgical management of colorectal lung metastasis. Clin Colon Rectal Surg 22, 233-241.
Vogel, T., Schumacher, V., Saleh, A., Trojan, J., and Moslein, G. (2000). Extraintestinal polyps in Peutz-Jeghers syndrome: presentation of four cases and review of the literature. Deutsche Peutz-Jeghers-Studiengruppe. Int J Colorectal Dis 15, 118-123.

115
Voloshanenko, O., Erdmann, G., Dubash, T.D., Augustin, I., Metzig, M., Moffa, G., Hundsrucker, C., Kerr, G., Sandmann, T., Anchang, B., Demir, K., Boehm, C., Leible, S., Ball, C.R., Glimm, H., Spang, R., and Boutros, M. (2013). Wnt secretion is required to maintain high levels of Wnt activity in colon cancer cells. Nat Commun 4, 2610.
Wagner, R.N., Proell, M., Kufer, T.A., and Schwarzenbacher, R. (2009). Evaluation of Nod-like receptor (NLR) effector domain interactions. PLoS One 4, e4931.
Walunas, T.L., Lenschow, D.J., Bakker, C.Y., Linsley, P.S., Freeman, G.J., Green, J.M., Thompson, C.B., and Bluestone, J.A. (1994). CTLA-4 can function as a negative regulator of T cell activation. Immunity 1, 405-413.
Wang, X., Herberg, F.W., Laue, M.M., Wullner, C., Hu, B., Petrasch-Parwez, E., and Kilimann, M.W. (2000). Neurobeachin: A protein kinase A-anchoring, beige/Chediak-higashi protein homolog implicated in neuronal membrane traffic. J Neurosci 20, 8551-8565.
Wasmuth, H.H., Trano, G., Myrvold, H.E., Aabakken, L., and Bakka, A. (2013). Adenoma formation and malignancy after restorative proctocolectomy with or without mucosectomy in patients with familial adenomatous polyposis. Dis Colon Rectum 56, 288-294.
Weber, J. (2010). Immune checkpoint proteins: a new therapeutic paradigm for cancer--preclinical background: CTLA-4 and PD-1 blockade. Semin Oncol 37, 430-439.
Weisenberger, D.J., Siegmund, K.D., Campan, M., Young, J., Long, T.I., Faasse, M.A., Kang, G.H., Widschwendter, M., Weener, D., Buchanan, D., Koh, H., Simms, L., Barker, M., Leggett, B., Levine, J., Kim, M., French, A.J., Thibodeau, S.N., Jass, J., Haile, R., and Laird, P.W. (2006). CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38, 787-793.
Willingham, S.B., Allen, I.C., Bergstralh, D.T., Brickey, W.J., Huang, M.T., Taxman, D.J., Duncan, J.A., and Ting, J.P. (2009). NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase-1 activation, necrosis, and HMGB1 release via inflammasome-dependent and -independent pathways. J Immunol 183, 2008-2015.
Win, A.K., Dowty, J.G., English, D.R., Campbell, P.T., Young, J.P., Winship, I., Macrae, F.A., Lipton, L., Parry, S., Young, G.P., Buchanan, D.D., Martinez, M.E., Jacobs, E.T., Ahnen, D.J., Haile, R.W., Casey, G., Baron, J.A., Lindor, N.M., Thibodeau, S.N., Newcomb, P.A., Potter, J.D., Le Marchand, L., Gallinger, S., Hopper, J.L., and Jenkins, M.A. (2011). Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer 105, 162-169.
Wing, K., Onishi, Y., Prieto-Martin, P., Yamaguchi, T., Miyara, M., Fehervari, Z., Nomura, T., and Sakaguchi, S. (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271-275.
Winkels, R.M., Botma, A., Van Duijnhoven, F.J., Nagengast, F.M., Kleibeuker, J.H., Vasen, H.F., and Kampman, E. (2012). Smoking increases the risk for colorectal adenomas in patients with Lynch syndrome. Gastroenterology 142, 241-247.
Woodhouse, E.C., Fisher, A., Bandle, R.W., Bryant-Greenwood, B., Charboneau, L., Petricoin, E.F., 3rd, and Liotta, L.A. (2003). Drosophila screening model for metastasis: Semaphorin 5c is required for l(2)gl cancer phenotype. Proc Natl Acad Sci U S A 100, 11463-11468.
Wu, J.S., Mcgannon, E.A., and Church, J.M. (1998). Incidence of neoplastic polyps in the ileal pouch of patients with familial adenomatous polyposis after restorative proctocolectomy. Dis Colon Rectum 41, 552-556; discussion 556-557.
Wu, X., Peng, M., Huang, B., Zhang, H., Wang, H., Xue, Z., Zhang, L., Da, Y., Yang, D., Yao, Z., and Zhang, R. (2013). Immune microenvironment profiles of tumor immune equilibrium and immune escape states of mouse sarcoma. Cancer Lett 340, 124-133.

116
Yan, H.H., Pickup, M., Pang, Y., Gorska, A.E., Li, Z., Chytil, A., Geng, Y., Gray, J.W., Moses, H.L., and Yang, L. (2010). Gr-1+CD11b+ myeloid cells tip the balance of immune protection to tumor promotion in the premetastatic lung. Cancer Res 70, 6139-6149.
Yang, H., Jeffrey, P.D., Miller, J., Kinnucan, E., Sun, Y., Thoma, N.H., Zheng, N., Chen, P.L., Lee, W.H., and Pavletich, N.P. (2002). BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure. Science 297, 1837-1848.
Yang, L., Huang, J., Ren, X., Gorska, A.E., Chytil, A., Aakre, M., Carbone, D.P., Matrisian, L.M., Richmond, A., Lin, P.C., and Moses, H.L. (2008). Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 13, 23-35.
Yokosuka, T., Kobayashi, W., Takamatsu, M., Sakata-Sogawa, K., Zeng, H., Hashimoto-Tane, A., Yagita, H., Tokunaga, M., and Saito, T. (2010). Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity 33, 326-339.
Zanke, B.W., Greenwood, C.M., Rangrej, J., Kustra, R., Tenesa, A., Farrington, S.M., Prendergast, J., Olschwang, S., Chiang, T., Crowdy, E., Ferretti, V., Laflamme, P., Sundararajan, S., Roumy, S., Olivier, J.F., Robidoux, F., Sladek, R., Montpetit, A., Campbell, P., Bezieau, S., O'shea, A.M., Zogopoulos, G., Cotterchio, M., Newcomb, P., Mclaughlin, J., Younghusband, B., Green, R., Green, J., Porteous, M.E., Campbell, H., Blanche, H., Sahbatou, M., Tubacher, E., Bonaiti-Pellie, C., Buecher, B., Riboli, E., Kury, S., Chanock, S.J., Potter, J., Thomas, G., Gallinger, S., Hudson, T.J., and Dunlop, M.G. (2007). Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat Genet 39, 989-994.
Zeisberg, E.M., Potenta, S., Xie, L., Zeisberg, M., and Kalluri, R. (2007). Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res 67, 10123-10128.
Zeisberg, M., Strutz, F., and Muller, G.A. (2000). Role of fibroblast activation in inducing interstitial fibrosis. J Nephrol 13 Suppl 3, S111-120.
Zhang, K., Civan, J., Mukherjee, S., Patel, F., and Yang, H. (2014). Genetic variations in colorectal cancer risk and clinical outcome. World J Gastroenterol 20, 4167-4177.
Zhang, Y., Cheng, S., Zhang, M., Zhen, L., Pang, D., Zhang, Q., and Li, Z. (2013a). High-infiltration of tumor-associated macrophages predicts unfavorable clinical outcome for node-negative breast cancer. PLoS One 8, e76147.
Zhang, Y., Davis, C., Ryan, J., Janney, C., and Pena, M.M. (2013b). Development and characterization of a reliable mouse model of colorectal cancer metastasis to the liver. Clin Exp Metastasis 30, 903-918.
Zhu, S., Barshow, S., Wildonger, J., Jan, L.Y., and Jan, Y.N. (2011). Ets transcription factor Pointed promotes the generation of intermediate neural progenitors in Drosophila larval brains. Proc Natl Acad Sci U S A 108, 20615-20620.
Zlobec, I., Bihl, M.P., Schwarb, H., Terracciano, L., and Lugli, A. (2010). Clinicopathological and protein characterization of BRAF- and K-RAS-mutated colorectal cancer and implications for prognosis. Int J Cancer 127, 367-380.
Zoratto, F., Rossi, L., Verrico, M., Papa, A., Basso, E., Zullo, A., Tomao, L., Romiti, A., Lo Russo, G., and Tomao, S. (2014). Focus on genetic and epigenetic events of colorectal cancer pathogenesis: implications for molecular diagnosis. Tumour Biol 35, 6195-6206.

117
FIGURES AND TABLES
Figure Legend
Figure 11. Role of innate immune effector proteins in CRC metastatic control. A) Survival of
Casp1-/-
(n = 9) , Casp12-/-
(n = 7) and WT ( n = 5) mice . Kaplan-Meier survival curve analysis
found no significant difference between Casp1-/-
and WT mice (p = 0.74), as well as no
significant difference between Casp12-/-
and WT mice (p = 0.82), following MC38met-Luc inv
tail vein injection. Graph represents one experiment. B) Survival of Ripk2
-/- (n = 9) and Birc3
-/-
(n = 5) compared to WT (n = 5; n = 5) mice following MC38inv tail vein injection. Kaplan-
Meier survival curve analysis found no significant difference between Ripk2-/-
and WT mice ( p =
0.18), as well as no significant difference between Birc3-/-
and WT mice (p = 0.56) following
MC38met-Luc inv tail vein injection. Graph represents one experiment. C) Survival Bid
-/- (n =
11) and Ripk3-/-
(n = 6) mice compared to WT (n = 6; n = 5) following MC38met-Luc inv tail
vein injection. Kaplan-Meier survival curve analysis found no significant difference between
Bid-/-
and WT mice ( p = 0.14), as well as no significant difference between Ripk3-/-
and WT
mice (p = 0.11) following MC38met-Luc inv tail vein injection. Data represents one experiment.
Figure 12. Presence of NK cells and ILCs correlates with increased resistance to MC38met-Luc
inv lung colonization. A) Survival of Jak3W81R
, Rag1-/-
and WT mice following MC38met-Luc
injection. Kaplan-Meier analysis of survival found a very significant difference in survival for
Rag1-/-
mice versus Jak3W81R
mice (p <0.0001) and WT mice versus Jak3W81R
mice (p<0.0001).
There was also a significant difference observed between Rag1-/-
and WT animals (p = 0.0094).
Results represent two individual experiments pooled (n = 10 per group in each experiment). B)
Lung weight of lungs in Jak3W81R
(n = 13), Rag1-/-
(n = 11) and WT (n = 13) mice dissected on
day 15 post-injection. One-way ANOVA analysis of results showed a significant difference in
the lung weights across all groups (p = 0.001) and a very significant difference in the lung
weights of WT and Jak3W81R
mice ( p < 0.01) and a significant difference between Jak3W81R
and
Rag1-/-
mice (p < 0.05). The graph is representative of three individual experiments. C) Histology
showing lung metastases (dark purple) of Jak3W81R
(n = 3), Rag1-/-
(n = 3) and WT (n = 3) mice
15 days post-injection and quantification of metastatic coverage on the right. T-tests between
groups found a significant difference in the percent lung metastatic coverage between WT and
Jak3W81R
animals (p = 0.044) and between Rag1-/-
and Jak3W81R
animals (p = 0.01). There was no

118
significant difference observed in the percent metastatic coverage between WT and Rag1-/-
animals (p = 0.98). Results are representative of two experiments. D) Bioluminescence of i.v.-
injected MC38met-Luc cells in vivo in Jak3W81R
(n = 10), Rag1-/-
(n = 10) and WT (n = 10) mice
as measured by a Xenogen IVIS 100 imaging system recorded every 5 days. T-test comparison
of the groups at individual timepoints found a significant difference between Jak3W81R
and
Rag1-/-
on Day 0 ( p = 0.0316), Day 10 (p = 0.0274 ) and Day 15 (p =0.0026 ), although no
significant difference on Day 5 ( p = 0.3956) . T-test comparison of Jak3W81R
and WT similarly
found significant differences in the signal measured for each group on Days 0 (p = 0.0022), 10 (
p = 0.0211) and 15 ( p = 0.0011), with no significant difference found on day 5 ( p = 0.0632). No
significant difference was recorded between WT and Rag1-/-
at any timepoint (Day 0, p = 0.3579;
Day 5, p = 0.3913; Day 10, p = 0.2989; Day 15, p = 0.1782; and Day 20, p = 0.4783). Jak3W81R
mice did not survive past day 15. Graph is representative of two individual experiments.
Figure 13. Antibody depletion of NK1.1+ and CD90.2
+ cells. A) Isolated lung cells from WT (n
= 10), Jak3W81R
(n = 9) and Rag1-/-
( n = 6) mice which were stained with an ILC and NK cell
Flow cytometry cocktail. The graph on the left represent the number of lung NK cells in each
animal, gated for live, lineage negative cells which are NK1.1+. The graph on the right
represents the number of ILCs found in each animal, gated for live, lineage negative cells which
are CD90.2+ and CD127+. One-way ANOVA analysis found a significant difference across all
groups for NK cells, with a significant difference in the number of NK cells between WT and
Jak3W81R
mice ( p < 0.01), WT and Rag1-/-
mice ( p < 0.001), as well as Rag1-/-
and Jak3W81R
mice ( p < 0.001). The data is representative of 2 individual experiments. B) NK1.1 cell gating
on a lineage negative population in a Rag1-/-
mouse and a Rag1-/-
mouse treated with the NK cell-
depleting antibody anti-GM1 15 days post MC38met-Luc injection. C) Quantification of the cell
population highlighted in red in B) is shown for PBS-treated Rag1-/-
( n = 6) anti-CD90-treated
Rag1-/-
( n = 7) and anti-GM1-treated Rag1-/-
(n = 6). One-way ANOVA analysis showed a
significant difference across all groups ( p < 0.0001) with a very significant difference between
PBS-treated and anti-CD90.2-treated Rag1-/-
( p < 0.001), PBS-treated and anti-GM1-treated
Rag1-/-
( p < 0.001), and anti-CD90.2-treated and anti-GM1-treated Rag1-/-
( p < 0.01).The data is
representative of two individual experiments. D) ILC cell gating on a lineage negative, CD90.2
positive and CD127 positive population in a Rag1-/-
mouse and a Rag1-/-
mouse treated with the
ILC-depleting antibody anti-CD90.2 15 days post MC38met-Luc injection. E) Quantification of

119
the cell population highlighted in red in D) is shown for PBS-treated Rag1-/-
( n = 6) anti-CD90-
treated Rag1-/-
( n = 7) and anti-GM1-treated Rag1-/-
(n = 6). One-way ANOVA analysis showed
a significant difference across all groups ( p < 0.0001) with a very significant difference between
PBS-treated and anti-CD90.2-treated Rag1-/-
( p < 0.001), PBS-treated and anti-GM1-treated
Rag1-/-
( p < 0.001), and no significant difference found between anti-GM1-treated and anti-
CD90.2-treated animals. The data is representative of two individual experiments.
Figure 14. NK cells contribute a more substantial role to protection than ILCs following
MC38met-Luc injection. A) Survival of Jak3W81R
(n = 5), PBS-treated Rag1-/-
( n = 5) anti-CD90-
treated Rag1-/-
( n = 4) and anti-GM1-treated Rag1-/-
( n = 4) mice following MC38met-Luc
injection. Kaplan-Meier analysis of survival found a significant difference in survival between
Jak3 and Rag1-/-
(p = 0.0027). Conversely, there was no significant difference found in the
survival of anti-CD90.2-treated Rag1-/-
and PBS-treated Rag1-/-
(p = 0.42) animals. There was
also a very significant difference reported in the survival of Jak3-/- and PBS-treated Rag1-/-
(p =
0.0016), Jak3 and CD90.2-treated Rag1-/-
(p = 0.002), anti-CD90.2-treated and anti-GM1-treated
Rag1-/-
(p = 0.0253) and PBS-treated and anti-GM1-treated Rag1-/-
animals (p = 0.0047). Results
are representative of one experiment. B) Lung weight of lungs in Jak3W81R
(n = 8), PBS-treated
Rag1-/-
(n = 6), anti-CD90-treated Rag1-/-
(n = 6) and anti-GM1-treated Rag1-/-
(n = 6) mice
dissected on day 15 post-injection. One-way ANOVA analysis of the results showed no
significant difference across groups (p = 6252), however a significant difference was found
between PBS-treated Rag1-/-
and anti-GM1-treated Rag1-/-
( p < 0.05). The graph represents two
individual experiments. C) Histology showing lung metastases (dark purple) of Jak3W81R
(n = 7),
PBS-treated Rag1-/-
(n = 6), anti-CD90-treated Rag1-/-
(n = 7) and anti-GM1-treated Rag1-/-
(n =
6) mice 15 days post-injection and quantification of metastatic coverage on the right. One-way
ANOVA analysis of the results showed a significant difference across all groups ( p = 0.034) and
a significant difference between PBS-treated Rag1-/-
and anti-GM1-treated Rag1-/-
( p < 0.05).
Results are representative of two individual experiments.
Figure 15. Summary of ENU screen and identified deviant pedigrees. A) Table summarizing
ENU screen. Over the course of the screen 500 G3 mice were screened from a total of 39 G1

120
males. Overall 2 families repeatedly displayed resistance to MC38met-Luc injection B)
Representation of a negative pedigree. Over the course of 4 MC38met-Luc injection rounds, 16
Family 38 animals were screened for differences in survival with injected WT and Jak3W81R
mice. Family 38 members all died within the same time frame as the accompanying WT, with
the exception of one animal in Round 3 which survived to 34 days, 7 days after the last WT
control died. This observation was never repeated however, and was likely caused by variability
in the model as opposed to a deviant phenotype induced by a genetic mutation. C) Pedigree 13;
x-axis lists individual injection rounds in which G3 mice and accompanying control animals
were injected with MC38met-Luc. G3 animals surviving past WT controls were considered
resistant. A total of 17 Family 13 members were screened over the course of 4 MC38met-Luc
injection rounds. No difference in survival compared to WT was observed in round 1, however,
the subsequent rounds 2, 3 and 4 repeatedly displayed animals surviving longer than WT at the
25% frequency expected when screening for a genetic candidate. Overall, 22% of animals (4
animals out of 18) displayed resistance, suggesting the possibility of a homozygous recessive
genotype responsible for the phenotype. Animals sent for exome sequencing are shown in the red
boxes. D) Pedigree 31; x-axis lists individual injection rounds in which G3 mice and
accompanying control animals were injected with MC38 inv. G3 animals surviving past WT
controls were considered resistant. A total of 25 animals were screened over the course of 6
injection rounds. In round 1, 2 out of 5 animals showed susceptibility following MC38met-Luc
injection compared to WT, but this pattern was not repeated in subsequent injection rounds. No
difference in survival compared to WT was observed in round 2, however, the subsequent rounds
3, 4, 5 and 6 repeatedly displayed animals surviving longer than WT at the 25% frequency
expected when screening for a genetic candidate. Overall, 20% of animals (5 animals out of 25)
displayed resistance, suggesting the possibility of a homozygous recessive genotype responsible
for the phenotype. Animals sent for exome sequencing are shown in the red boxes.
Figure 16. Candidate Mutation found in 4930430F08Rik. Mutation in exon 5 of 4930430F08Rik
replacing guanine for adenosine, resulting in a serine to leucine amino acid.
Figure 17. Candidate Mutation found in Ap1g2. A) Mutation in exon 14 of Ap1g2 replaces
cytosine for adenosine, resulting in an B) arginine to leucine amino acid change. C) Conservation
of the protein at this position can be seen in several species.

121
Figure 18. Candidate Mutation found in A130010J15Rik. A) Mutation in exon 2 of
A130010J15Rik replaces a thymine for acytosine, resulting in a B) valine to alanine amino acid
change.
Figure 19. Candidate Mutation found in Nbeal1. A) Mutation in exon 35 of Nbeal1 replaces an
adenosine for a guanine, resulting in a B) serine to glycine amino acid change. C) Conservation
of the protein at this amino acid postion can be across species.
Figure 20. Candidate Mutation found in Cadm3. A) Mutation in exon 8 of Cadm3 replaces a
cytosine for an adenosine, resulting in an B) alanine to serine amino acid change. C)
Conservation of the protein at this amino acid position can be seen across species.
Figure 21. Candidate Mutation found in Ripk2. A) Mutation in intron 10 of Ripk2 replaces an
adenosine for a guanine, resulting in a change in splicing of the mRNA transcript.
Table 2. Family 13 gene candidate mutations following exome sequencing.
Table 3. Family 31 gene candidate mutations following exome sequencing.
Table 4. Expression of candidate genes in human normal and cancerous tissue.
122
Figure 11.
N = 6
N = 5
N = 9
N = 5
N = 5
N = 5
N = 9
N = 7
N = 11
N = 6 N = 5

123
Figure 12.

124
Figure 13.

125
Figure 14.
6 mm
Jak3W81R Rag1-/- PBS Rag1-/- anti CD90 Rag1-/- anti GM1
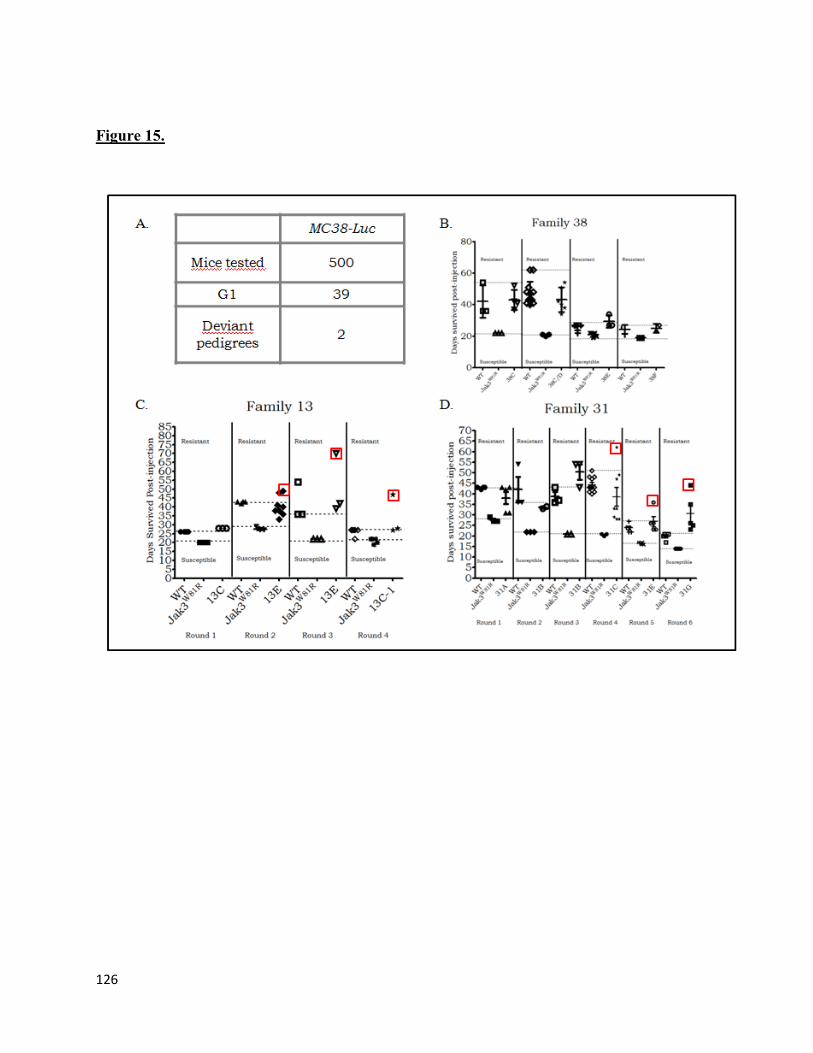
126
Figure 15.

127
Figure 16.
Figure 17.

128
Figure 18.
Figure 19.

129
Figure 20.
Figure 21.

130
Table 2.

131
Table 3.
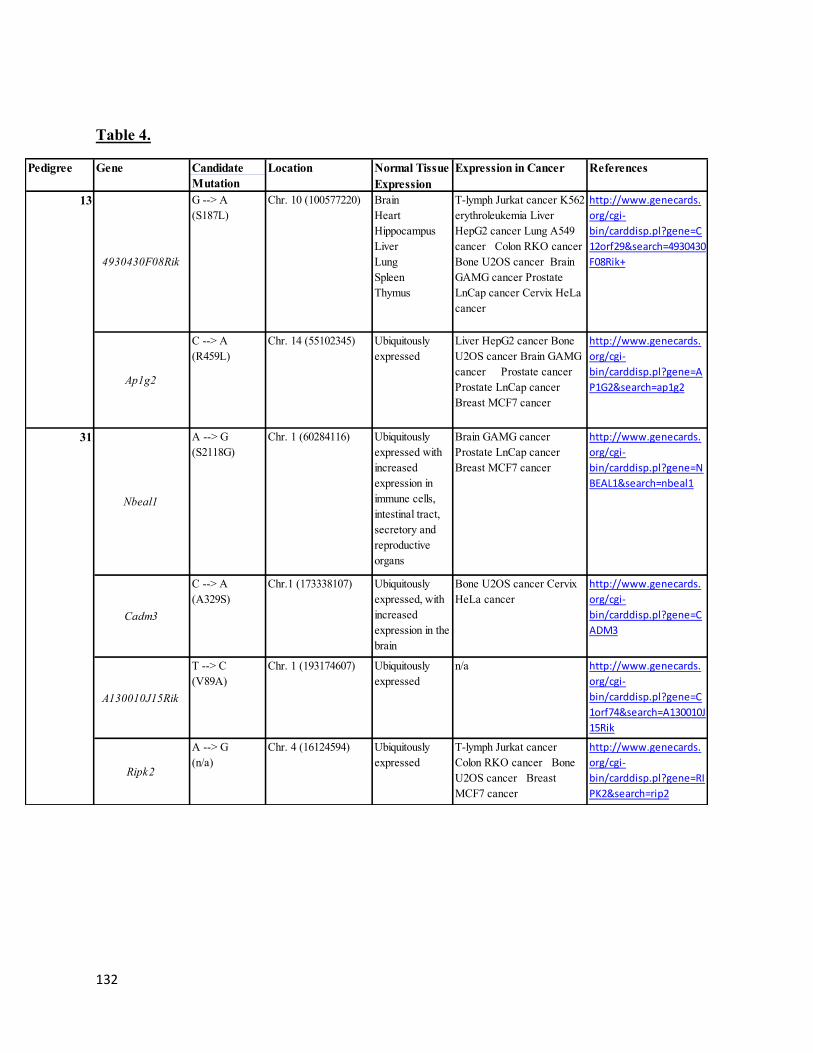
132
Table 4.
Candidate
Mutation
Brain GAMG cancer
Prostate LnCap cancer
Breast MCF7 cancer
Chr. 1 (60284116)
Bone U2OS cancer Cervix
HeLa cancer
Chr.1 (173338107)
Ubiquitously
expressed
http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=RI
PK2&search=rip2
Chr. 1 (193174607)
T-lymph Jurkat cancer
Colon RKO cancer Bone
U2OS cancer Breast
MCF7 cancer
Chr. 4 (16124594)
31
Nbeal1
A --> G
(S2118G)
Ubiquitously
expressed with
increased
expression in
immune cells,
intestinal tract,
secretory and
reproductive
organs
http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=N
BEAL1&search=nbeal1
Cadm3
C --> A
(A329S)
Ubiquitously
expressed, with
increased
expression in the
brain
http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=C
ADM3
A130010J15Rik
T --> C
(V89A)
Ubiquitously
expressed
n/a http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=C
1orf74&search=A130010J
15Rik
Ripk2
A --> G
(n/a)
13
4930430F08Rik
G --> A
(S187L)
http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=C
12orf29&search=4930430
F08Rik+
Ap1g2
C --> A
(R459L)
Ubiquitously
expressed
http://www.genecards.
org/cgi-
bin/carddisp.pl?gene=A
P1G2&search=ap1g2
T-lymph Jurkat cancer K562
erythroleukemia Liver
HepG2 cancer Lung A549
cancer Colon RKO cancer
Bone U2OS cancer Brain
GAMG cancer Prostate
LnCap cancer Cervix HeLa
cancer
Brain
Heart
Hippocampus
Liver
Lung
Spleen
Thymus
Chr. 10 (100577220)
Liver HepG2 cancer Bone
U2OS cancer Brain GAMG
cancer Prostate cancer
Prostate LnCap cancer
Breast MCF7 cancer
Chr. 14 (55102345)
ReferencesPedigree Gene Location Normal Tissue
Expression
Expression in Cancer