metagenomic analysis of kimchi, the korean traditional ... · 2 25 26 kimchi, the korean...
TRANSCRIPT

1
Metagenomic Analysis of Kimchi, the Korean Traditional 1
Fermented Food 2
3
Ji Young Jung1, Se Hee Lee
1, Jeong Myeong Kim
1, Moon Su Park
1, Jin-Woo Bae
2, Yoonsoo 4
Hahn1, Eugene L. Madsen
3 and Che Ok Jeon
1,* 5
6
1Schools of Biological Sciences, Research Center for Biomolecules and Biosystems, Chung-7
Ang University, Seoul, 156-756, Republic of Korea 8
2Department of Biology, Kyung Hee University, Seoul, 130-701, Republic of Korea 9
3Department of Microbiology, Cornell University, Ithaca, NY, 14853-8101, U.S.A. 10
11
12
Running title: Microbiome of kimchi 13
14
*Corresponding author: Che Ok Jeon. 15
Mailing address: Department of Life Science, Chung-Ang University, 221, HeukSeok-Dong, 16
Seoul, 156-756, Republic of Korea. 17
Tel: +82-2-820-5864. 18
Fax: +82-2-821-8132. 19
E-mail: [email protected] 20
21
Subject Category: microbial population and community ecology 22
23
Key words: kimchi, metagenomics, lactic acid bacteria, pyrosequencing, fermented food 24
Copyright © 2011, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved.Appl. Environ. Microbiol. doi:10.1128/AEM.02157-10 AEM Accepts, published online ahead of print on 11 February 2011
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

2
25
Kimchi, the Korean culture's traditional food, is made from vegetables by fermentation. 26
In this study, metagenomic approaches were used to monitor changes in bacterial 27
populations, metabolic potential, and overall genetic features of the microbial 28
community during the 29-day fermentation. Metagenomic DNA was extracted from 29
kimchi samples obtained periodically and followed by sequencing using a 454 GS-FLX 30
Titanium system, which yielded a total of 701,556 reads, with an average read length of 31
438 bp. Phylogenetic analysis based on 16S rRNA genes from the metagenome indicated 32
that the kimchi microbiome was dominated by members of three genera; Leuconostoc, 33
Lactobacillus, and Weissella. Assignment of metagenomic sequences to SEED categories 34
of MG-RAST revealed a genetic profile characteristic of heterotrophic lactic acid 35
fermentation of carbohydrates, which was supported by the detection of mannitol, 36
lactate, acetate, and ethanol as fermentation products. When the metagenomic reads 37
were mapped onto the database of completed genomes, the Leuconostoc mesenteroides s38
ubsp. mesenteroides ATCC 8293 and Lactobacillus sakei subsp. sakei 23K genomes 39
were highly represented. These same two genera were confirmed to be important in 40
kimchi fermentation when the majority of kimchi metagenomic sequences showed very 41
high identity to Leuconostoc mesenteroides and Lactobacillus genes. Besides microbial 42
genome sequences, a surprisingly large number of phage DNA sequences were identified 43
from the cellular fractions, possibly indicating that a high proportion of cells were 44
infected by bacteriophage during fermentation. Overall, these results provide insights 45
into the kimchi microbial community and also shed light on fermentation processes 46
carried out broadly by complex microbial communities. 47
48
49
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

3
Kimchi is a traditional food, emblematic of Korean culture, that is fermented from vegetables 50
such as Chinese cabbage and radish. In recent years, kimchi's health-promoting characteristics 51
have been recognized (40, 54). Kimchi is usually processed with various seasoning 52
ingredients such as red pepper powder, garlic, ginger, green onion, fermented seafood 53
(jeotgal), and salts at low temperatures to ensure proper ripening and preservation. 54
Spontaneous fermentation without the use of starter cultures or sterilization leads to the 55
growth of various microorganisms during kimchi preparation. Moreover, the influence of 56
ingredients and fermentation conditions on the microbial community are unexplored, and a 57
rational approach to control of the microbial community for improvement of kimchi taste 58
(flavor) is currently almost impossible. 59
Microbes involved in kimchi fermentation have been studied previously (26, 27). 60
Taxonomic studies of bacteria isolated and purified from kimchi fermenting cultures have 61
shown that lactic acid bacteria (LAB) including Leuconostoc (Le.) mesenteroides, Le. kimchii, 62
Le. citreum, Le. gasicomitatum, Le. gelidum, Lactobacillus (Lb.) brevis, Lb. curvatus, Lb. 63
plantarum, Lb. sakei, Lactococcus lactis, Pediococcus pentosaceus, Weissella (W) confusa, W. 64
kimchii, and W. koreensis are likely to be key players responsible for kimchi fermentation (2, 65
22, 24, 28, 42, 51). However, culture-based approaches have known limitations in terms of 66
reproducibility and challenges in the culturability of some bacteria. Therefore, the set of 67
cultured isolates may not reflect the true microbial composition of fermented kimchi. 68
The use of 16S rRNA genes as a phylogenetic marker that does not require isolation and 69
cultivation of microorganisms has resulted in a tremendous amount of information about 70
naturally occurring microbial communities in various habitats (9, 48). When applied to kimchi, 71
the construction of 16S rRNA clone libraries has extended information about kimchi 72
microbiology (42). Denaturing gradient gel electrophoresis and amplified ribosomal DNA 73
restriction analysis, followed by sequencing of 16S rRNA genes, were additional techniques 74
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

4
used to elucidate microbial community involved in kimchi fermentation (24, 28). Although 75
these culture-independent approaches can estimate species richness within a kimchi microbial 76
community, they are unable to resolve the true genotypic diversity because diversity of 16S 77
rRNA gene sequences may not correlate well with genotypic and phenotypic diversity (60). 78
Thus, although progress using 16S RNA gene-analysis and culture-based methods has been 79
significant, current understanding of kimchi microbiology is piecemeal and features 80
significant limitations regarding community dynamics and function during fermentation (12, 81
29, 38). 82
Metagenomic approaches based on the random sequencing of environmental DNA can 83
provide a wealth of information (free of PCR bias) about gene content, metabolic potential, 84
and the function of microbial communities (10). Recently, the development of next-generation 85
DNA sequencing technologies (50), such as 454-pyrosequencing has led large-scale 86
metagenomic sequencing of environmental habitats such as activated sludge (16, 49), coastal 87
ocean (36), glacier ice (52), and the human distal gut (45). 88
It is well known that kimchi fermentation is generally characterized by the production of 89
metabolites such as organic acids (lactic and acetic acids) and other flavoring compounds, 90
which are important components in defining kimchi quality (20). The presence of various 91
metabolites can reflect the presence of related genes in the microbial population, and give a 92
more direct collective phenotypic view of the kimchi microbiome as a consequence of the end 93
products of gene expression. Fermentation rate is also very important to kimchi taste and 94
flavor development during fermentation (25). 1H nuclear magnetic resonance (NMR) is one of 95
the most adequate, easy, and nondestructive multinuclear techniques to simultaneously 96
monitor several substances present in one sample (15, 30). 97
Here, we applied metagenomic approaches using pyrosequencing technology developed by 98
454 Life Sciences to kimchi. In addition, we conducted a metabolite analysis of fermenting 99
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

5
kimchi using 1H NMR to collectively understand the overall features of the kimchi 100
microbiome and to investigate relationships between metabolite production, metabolic 101
potential, and composition of the kimchi microbial community during fermentation. 102
103
MATERIALS AND METHODS 104
Preparation of kimchi and sampling. An industrial scale batch of kimchi was prepared by 105
Daesang F&F Corporation (http://www.chonggafood.com) using their production line. Briefly, 106
Chinese cabbage (Brassica rapa subsp. pekinensis) were soaked in a solution of 15% (w/v) 107
solar salt for 5 to 6 h. The soaked Chinese cabbages were washed with tap water 3 times and 108
drained excessive water. The salted Chinese cabbages were mixed with various seasoning 109
ingredient mixtures including red pepper powder, radish, garlic, ginger, green onion, sugar, 110
and fermented seafood (jeotgal) and were packaged into airtight plastic bags with 1-kg 111
portions. Thirty 1 kg kimchi bags were stored at the standard fermentation temperature (4°C). 112
Periodically over the 29-day fermentation period, 3 bags were sacrificed and subjected to 3 113
layers of coarse gauze (Daehan, Korea) to remove only large kimchi particles and the filtrates 114
were pooled. The pH of each filtrate was measured immediately and the filtrates were 115
centrifuged (8,000 rpm for 20 min at 4°C) to harvest microorganisms. The pellets and 116
supernatants were stored at -80°C for extraction of bulk genomic DNA and metabolite 117
analysis, respectively. One ml of each sample was centrifuged separately for the measurement 118
of 16S rRNA gene copies using quantitative real time-PCR (qRT-PCR). The kimchi samples 119
were coded as J"x", where "x" designates the sample day during fermentation. 120
121
Quantitative real time-PCR (qRT-PCR) to determine 16S rRNA gene copy number. To 122
estimate the total number of bacteria during the kimchi fermentation, total genomic DNA 123
from pellets derived from 1.0 ml of kimchi filtrate by centrifugation was extracted using a 124
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

6
FastDNA SPIN Kit (MPbio, Solon, OH, USA) according to manufacturer's instructions. 125
Genomic DNA concentrations were measured using an ELISA reader equipped with Take3TM
126
Multi-Volume Plate (SynergyMx, BioTek, CA, USA). The genomic DNA was serially diluted 127
ranging from 50 µg/ml ~ 200 µg/ml and was used as template to amplify 16S rRNA genes 128
from each group of bacteria using primers, 340F: 5'-CCTACGGGAGGCAGCAG-3' and 129
758R: 5'-CTACCAGGGTATCTAATCC-3' (21). The PCR mixture consisted of 2 µl of 130
genomic DNA, 0.3 µM total of the primer set (340F, 758R), 10 µl of 2X iQTM
SYBR®
Green 131
Supermix (Bio-Rad, Hercules, CA, USA), and water to a total of 20 µl. The qRT-PCR 132
amplifications were conducted using the CFX96TM
Real-Time PCR system (Bio-Rad, 133
Hercules, CA, USA) with the following qRT-PCR conditions: initial denaturation at 95°C for 134
3 min; followed by 40 cycles of 95°C for 45 sec, 59°C for 45 sec, and 72°C for 50 sec. 135
Bacterial standard curves were generated on the basis of the concentration of a pCR2.1 vector 136
(Invitrogen, CA, USA) carrying the 16S rRNA gene of Leuconostoc mesenteroides subsp. 137
mesenteroides ATCC 8293T. The 16S rRNA gene copy number of each sample was calculated 138
as described previously (41, 46). 139
140
1H-NMR spectroscopic analysis of kimchi metabolites. The analysis of metabolites, 141
including organic acids and monosaccharides in kimchi supernatants during fermentation, was 142
performed using 1H-NMR spectroscopy according to the modification of the methods 143
described previously (30, 53). Briefly, 15 ml of kimchi supernatant without cells was adjusted 144
to pH 6.0 and then lyophilized. The freeze-dried powders were dissolved in 600 µl of D2O 145
(10%, w/v) with 0.5 mM sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS) and 146
centrifuged at 13,000 rpm for 5 min. The resulting supernatants were transferred into 5 mm 147
NMR tubes and 1H-NMR spectra were acquired using a Varian Inova-600 MHz NMR 148
spectrometer (Varian Inc., Palo Alto, CA). Identification and quantification of individual 149
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

7
metabolites was performed using the Profiler module of the Chenomx NMR Suite v. 6.1 150
(Chenomx. Inc., Edmonton, Canada). 151
152
Genomic DNA extraction and pyrosequencing. Total genomic DNA from pellets was 153
extracted using a FastDNA SPIN Kit (MPbio) according to manufacturer's instructions. The 154
extracted crude DNA was loaded on 1% (w/v) low-melting agarose (Promega, WI, USA) and 155
electrophoresed at 100 V for 1 hr to remove impurities. A strip from each side of the gel was 156
removed and stained to localize the genomic DNA. DNA-containing regions from the rest of 157
the unstained gel were removed, digested with β-agarase I, and DNA recovery was conducted 158
as described by the manufacturer (NEB, MA, USA). The concentration of purified genomic 159
DNA was measured with an ELISA reader equipped with Take3TM
Multi-Volume Plate 160
(SynergyMx, BioTek, CA, USA) and the DNA samples were concentrated to a minimum of 161
100 ng/µl using a speed vacuum centrifuge (EYELA CVE-2000, Japan). The total genomic 162
DNA was nebulized to obtain a mean fragment size of ~680 bp, confirmed by electrophoresis, 163
for pyrosequencing. The fragmented genomic DNA was individually tagged using multiplex 164
identifier (MID) adaptors according to the manufacturer’s instructions (Roche, Mannheim, 165
Germany), which allowed sorting of the pyrosequencing-derived sequencing reads based on 166
MID adaptors automatically after pyrosequencing. Pyrosequencing of the 10 tagged genomic 167
DNA samples was performed using the 454 GS-FLX Titanium system (Roche, Mannheim, 168
Germany) by Macrogen (Korea). 169
Sequencing data sets were subjected to systematic checks to remove replicates, duplicates, 170
and low quality reads. Briefly, replicate and duplicate sequences, which are known artifacts of 171
the pyrosequencing approach, were first checked through a web based program 172
(http://microbiomes.msu.edu/replicates/) (18) using the default settings (sequence identity 173
cutoff: 0.9; length identity requirement: 0; number of beginning base pairs to check: 3) of the 174
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

8
open-source program Cd-hit (31). Replicates and duplicates were removed, and the remaining 175
sequence data were then trimmed using the LUCY program (7) with a stringency similar to 176
Phred scores of 20 or higher (-error 0.01), and long reads over 100 bp. Only high quality 177
sequence data obtained after LUCY trimming were used for further subtraction with SeqClean 178
(http://compbio.dfci.harvard.edu/tgi/software/). Resulting clean sequence data sets were then 179
used for further analysis. 180
181
Comparative analysis of metagenomic data. Unassembled clean reads were annotated using 182
the MG-RAST server, an open-access metagenome curation and analysis platform 183
(http://metagenomics.nmpdr.org/) (35). To analyze bacterial community changes during 184
kimchi fermentation, 16S rRNA gene sequences from all sequences were identified by 185
comparing the read data set against the RDP II database (8), using recommended parameters 186
(minimum alignment length ≥ 50 bp, E-value = 0.01 cutoffs). 16S rRNA gene phylotypes 187
were classified at the genus, family, order, class, and phylum-level based on the taxonomic 188
categorizations of the MG-RAST server, respectively. To identify putative protein-encoding 189
sequences, the sequences were also analyzed using the SEED platform 190
(http://www.theseed.org/), which comprises all available genomic data from genome 191
sequencing centers (1). Kimchi microbial community metabolic potential was determined by 192
assigning functional annotation to metagenomic sequences with subsequent sequence 193
assignment to SEED subsystems using the recommended parameters (minimum alignment 194
length ≥50 bp, E-value = 0.01 cutoffs). Statistical analyses were conducted on the number of 195
sequencing reads within each subsystem rather than on the number of total metagenomic 196
reads. Therefore, abundance of metabolic genes in the MG-RAST server was calculated based 197
on the relative proportion of sequencing reads within each subsystem. Metabolic potential of 198
the kimchi microbiome was compared with other metagenomic data sets of the MG-RAST 199
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

9
server. 200
201
Individual genome recruitment plots. The degree to which completed, whole-genome 202
sequences were represented in metagenomic reads was assessed using the "recruitment plot" 203
procedure of Ghai et al., (17). Sequence fragments from unassembled clean reads derived 204
from the 454 GS-FLX Titanium system were compared to all complete and draft microbial 205
genomes using the BLASTN algorithm. Whole clean metagenomic reads were matched to 206
multiple reference genomes and were matched multiple times to the same genome (17). The 207
criteria for counting a given sequence were a minimum percent identity of 95% and a 208
minimum alignment of 90 bp. Whole clean reads were multiply-matched to the four highest 209
scoring genomes: Leuconostoc mesenteroides subsp. mesenteroides ATCC 8293T 210
(NC_008531), Lactobacillus sakei 23K sakei (NC_007576), the partially assembled genome 211
of Weissella paramesenteroides ATCC 33313, and Leuconostoc citreum KM20 (NC_010471). 212
The data were plotted using the plotting program gnuplot (http://www.gnuplot.info/). 213
214
Assembly of pyrosequencing-derived reads and analysis of assembled contigs. 215
Pyrosequencing-derived sequencing reads were assembled using the 454 Newbler Assembler 216
(ver. 2.0.01.14, Roche) by Macrogen (Korea). Low complexity sequence regions (simple 217
sequence repeats) were identified and excluded from consideration during initial pair-wise 218
comparison, but were included during final alignment and consensus building. Assembled 219
contigs longer than 10 kb were assigned to their taxonomic affiliations by BLASTN 220
comparisons to the GenBank nonredundant nucleotide (nr/nt) database and selections of the 221
top BLASTN hits for phylogenetic affiliation as described previously (43, 44). Clustered 222
regularly interspaced short palindromic repeats (CRISPR) elements were searched from 223
assembled contigs longer than 10 kb using a web-based CRISPRFinder (19). 224
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

10
225
Phylogenetic analysis of phage-related contigs and analysis of their abundance. Four 226
contigs (contig01645, contig27827, contig02778, and contig03545), assigned as putative 227
bacteriophage genome fragments among the assembled contigs, were analyzed in detail. 228
Molecular phylogenetic analysis of the putative phage contigs was performed using 229
bacteriophage major capsid proteins (MCPs). Putative MCP sequences were aligned with 230
homologous MCP sequences and retrieved from the NCBI database using the MUSCLE 231
program (13). Phylogenetic trees were constructed using MEGA (ver. 4.0) based on deduced 232
amino acid sequences (55). The abundance of putative phage reads in the cellular fractions of 233
kimchi supernatants during fermentation was estimated by multiple matching of the 234
metagenomic data sets against the four putative phage contigs, with the criteria of a minimum 235
percent identity of 95% and a minimum overlap of 90 bp as individual genome recruitment 236
plots were performed above. 237
238
Nucleotide accession numbers. The kimchi metagenomic data sets are publicly available in 239
the MG-RAST system under the following project identifiers (IDs 4450209.3 ~ 4450218.3) 240
and the NCBI Short Read Achieve (Accession # :SRA023444). Assembled contig sequences 241
with more than 10 kb have been deposited in GenBank (AEKF01000001 ~ AEKF01000127). 242
243
RESULTS AND DISCUSSION 244
Metagenomics makes it possible to study microbial communities by deciphering genetic 245
information from DNA extracted directly from naturally occurring habitats. Various 246
metagenomic approaches have been applied to environmental samples to characterize the 247
microbial and viral communities present in an ecosystem, thereby elucidating metabolic 248
capabilities and native taxa (11, 47, 56, 57, 59). Advantages of metagenomic studies include 249
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

11
avoiding the need to culture individual microorganisms, elimination of cloning and PCR 250
biases, and reduced cost per sequencing read. At present, the 454 GS-FLX Titanium platform 251
generates relatively long read lengths (400~500 bp), which makes it possible to reliably 252
assemble metagenomic reads. Here, we applied pyrosequencing technology to analyze the 253
microbiome of kimchi, the most popular fermented food in Korea. In addition, we analyzed 254
the metabolites of fermenting kimchi using 1H NMR to investigate the link between kimchi 255
microbial community metagenome and phenotype. 256
257
General features of kimchi fermentation. Kimchi fermentation is an anaerobic process, and 258
sampling kimchi supernatants from one large fermentation batch can cause oxygen infiltration, 259
disturbing the normal fermentation process and thereby influencing the microbial community 260
and metabolite production. Therefore, thirty sealed bags, each containing 1 kg of kimchi that 261
was manufactured by a one-batch process (incubated at the standard 4°C temperature), were 262
prepared for metagenome and metabolite analysis. The pH profile of kimchi supernatants over 263
the 30 day-fermentation process (Fig. 1) was similar to those of typical kimchi fermentation 264
(41). In this study, the pH was about 5.5 at the beginning and decreased sharply after 16 days. 265
After 27 days of fermentation, the pH reached below 4.5 and became stable. 266
A qRT-PCR approach was used to enumerate the total number of bacteria present during 267
the kimchi fermentation process. The exact determination of bacterial cell number by qRT-268
PCR in a microbial community is impossible because the copy number of chromosomal 16S 269
rRNA gene operons varies with species type (14); however, the data does allow for an 270
estimation of cell number. In our study, the total number of bacteria in kimchi samples was 271
estimated using a standard curve (coefficients (R2) value= 0.999) prepared from the 16S 272
rRNA gene of Leuconostoc mesenteroides subsp. mesenteroides ATCC 8293. The 16S rRNA 273
gene copy numbers of kimchi samples increased from an initial value (sample J1, day 1) of 274
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

12
2.6 × 108
copies/ml to the highest value (sample J25, day 25) of 3.0 × 1010
copies/ml during 275
the kimchi fermentation. After the day 25 sample, the bacterial population decreased steadily. 276
The increase of bacterial abundance correlated well with a decrease in pH value (Fig. 1). 277
278
Identification and quantification of metabolites during the kimchi fermentation. It is 279
known that kimchi flavor is principally dependent on free sugars, amino acids, and organic 280
acids, production of which are surely influenced by the microbial community during 281
fermentation (20). Therefore, we analyzed the change of metabolites and microbial 282
communities during kimchi fermentation using 1H-NMR spectroscopy to investigate their 283
relationships. Identification and quantification of metabolites using 1H-NMR peaks were 284
performed based on comparison with chemical shifts of standard compounds using Chenomx 285
NMR suite software. 1H-NMR spectra of the kimchi supernatants were associated with the 286
presence of various metabolites, including carbohydrates, amino acids, and organic acids as 287
expected. In particular, intense parts of the 1H-NMR spectra were in the 0.5-2.0 ppm and 3.0-288
5.0 ppm ranges, corresponding to organic acids and carbohydrates, respectively (data not 289
shown). 290
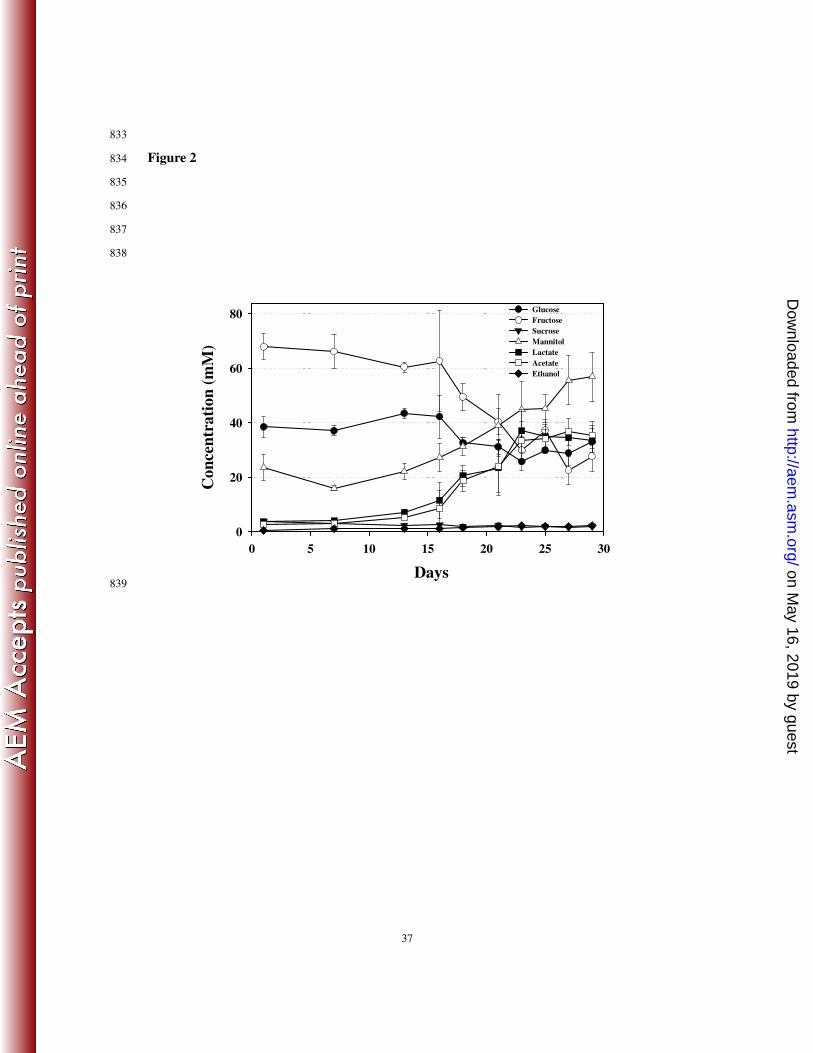
In this study, fructose and glucose were detected as major free sugars (Fig. 2). Free sugars 291
play important roles in the taste of kimchi because free sugars are not only sweeteners but 292
also serve as carbon sources for microorganisms, including LAB, to produce various products. 293
The level of free sugars decreased slowly in the early phases; between days 16 and 23, their 294
concentrations decreased relatively quickly. After day 23, carbohydrate levels were relatively 295
constant (Fig. 2). With the decrease in free sugars, lactate, acetate, and ethanol were detected 296
as fermentation products, indicating that heterotrophic lactic acid fermentation occurred 297
during fermentation. It was also found that a considerable amount of mannitol accumulated 298
during the fermentation (Fig. 2), which is a naturally occurring six-carbon polyol produced 299
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

13
from the reduction of fructose by LAB in vegetable fermentations (5, 61). The presence of 300
mannitol in foods results in a refreshing taste with tooth-friendly properties and is a good 301
replacement for sugars in diabetic foods (61). In general, the profile of fermentation products 302
inversely correlated with the level of free sugars. However, the level of fermentation products, 303
especially mannitol, increased continually at the early and late phases, and free sugars were 304
relatively constant, which might be caused by the continuous liberation of free sugar from 305
cabbage. The change in free sugars and fermentation products were closely correlated with 306
pH change and growth of the bacterial population (Fig. 1). However, ethanol levels were very 307
low, which might be explained by the loss of ethanol during sample preparation (freeze 308
drying) for NMR analysis. 309
310
Statistics of sequences generated by 454-pyrosequencing. Pyrosequencing of ten kimchi 311
samples during fermentation yielded a total of 701,566 reads by single run of 10 tagged 312
samples (Table 1). However, there was considerable variation in the number of sequences 313
obtained from each 454 sequencing sample, which could be explained by a difference in the 314
amount of genomic DNA from each sample, even though the total amount of DNA was 315
adjusted before pooling for sequencing. It is well known that the 454 sequencers produce 316
artificial replicated and duplicated reads, which might produce misleading conclusions (18). 317
Therefore, replicates, duplicates, low quality and short read length sequences (<100 bp) were 318
removed from the raw data set through web-based approaches. Here, these culled sequences 319
constituted 18.9~21.3% depending on the kimchi sample, which was a typical output for the 320
Roche GS-FLX system (39). Finally, 560,907 clean sequencing reads (79.95% of total reads) 321
with an average sequence length of 438 bp were obtained. This allowed clear assembly and 322
annotation analysis. The G+C% content of the data sets ranged from 30~40%, which reflects 323
the predominance of low G+C Gram-positive LAB. Table 1 summarizes the statistics for 324
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

14
pyrosequencing-derived data sets from the kimchi samples. 325
326
Phylogenetic compositions of the bacterial communities. Among the pyrosequencing-327
derived data sets, 16S rRNA genes accounted for 0.17~0.67% with an average of 0.54% 328
(Table 1). The number of 16S rRNA genes increased as the fermentation progressed and the 329
16S rRNA gene frequencies were slightly higher than in cultured genome prokaryotic 330
genomes (36); perhaps reflecting kimchi’s enrichment in LAB, which contain several copies 331
of 16S rRNA genes within relatively small genomes (~2 Mb). Sequences were classified at 332
each level using the MG-RAST server RDP (16S rRNA gene) database (E-value = 0.01 using 333
a minimum alignment length ≥ 50 bp). Phylogenetic classifications of the 16S rRNA gene 334
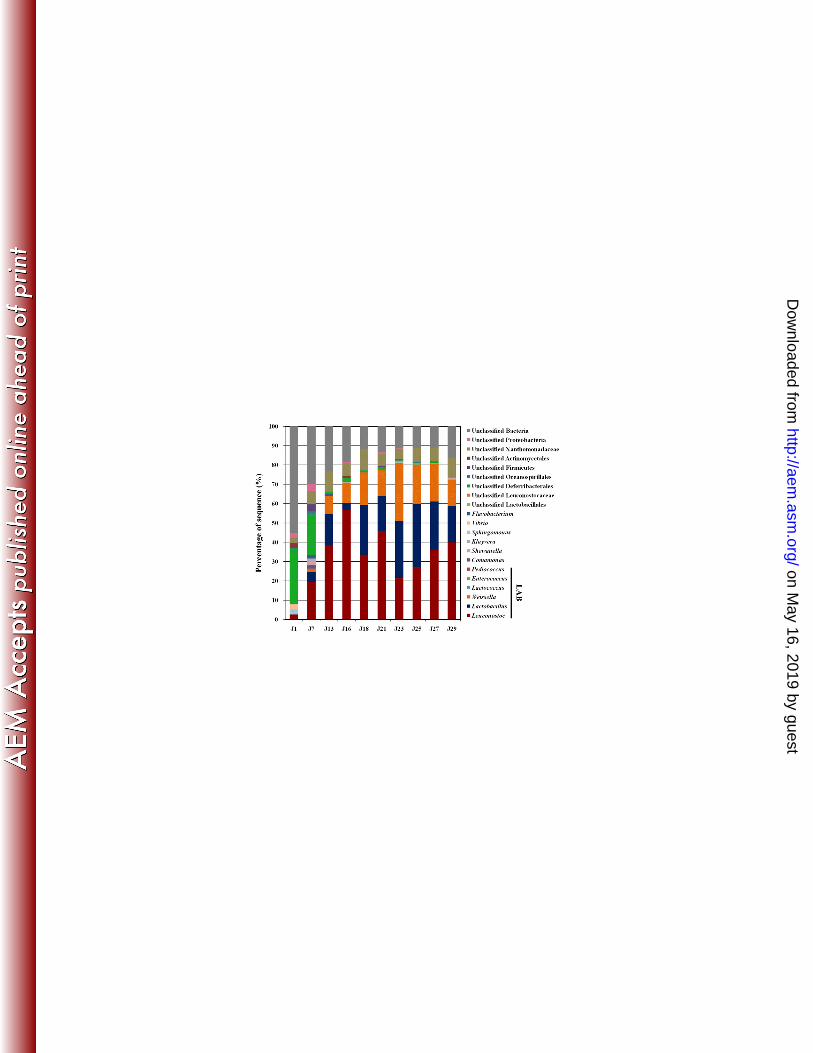
sequences are shown in Fig. 3. 335
Genomic DNA from kimchi substrates such as Chinese cabbage, red pepper, garlic, and 336
fermented seafood could be a reason for the relatively low frequency of 16S rRNA genes in 337
the early fermentation phases (J1 = 0.17%, J7 = 0.19%). In fact, in early fermentation the 338
proportion of unclassified phylotypic groups including unclassified Bacteria and unclassified 339
Deferribacterales were highest (J1 = ~97%), and they might have originated from Chinese 340
cabbage and kimchi additives. However, the proportion of unclassified phylotypic groups 341
decreased gradually as the fermentation progressed due to an increase in bacterial abundance. 342
Over the entire kimchi fermentation, somewhat greater 20% of the 16S rRNA gene sequences 343
for all samples fell into unclassified phylotypic groups. Archaeal 16S rRNA gene sequences 344
were not detected. 345
Figure 3 shows that the kimchi fermentation was governed by the distinct population 346
dynamics of three genera, Leuconostoc, Lactobacillus and Weissella. Among them, the genus 347
Leuconostoc was most abundant over the kimchi fermentation, followed by Lactobacillus and 348
Weissella. Leuconostoc dominated at the beginning stages of kimchi fermentation, but as the 349
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

15
fermentation progressed the abundance of Lactobacillus and Weissella increased. After the 350
middle stages of fermentation, Leuconostoc, Lactobacillus and Weissella became the 351
predominant bacterial groups in the microbial community and their cumulative abundance 352
reached to approximately 80% after day 23 (sample J23), which comprised more than 98% of 353
all bacterial groups, except for unclassified phylotypic groups. However, after day 23, the 354
abundance of Leuconostoc increased again, and Lactobacillus and Weissella gradually 355
decreased. 356
Previous studies reported that in kimchi fermentation, Leuconostoc species typically 357
dominate during the beginning of fermentation with low acidity at high temperature, whereas 358
Weissella and Lactobacillus species have the capacity to grow under high acidity and low 359
temperature, indicating that fermentation temperature and acidity are important determinants 360
of the microbial population (6, 28, 29). However, our results showed that Leuconostoc species 361
dominated, even at low temperature (4°C) and high acidity. Although Leuconostoc species 362
were more dominant than Weissella and Lactobacillus species at the beginning of 363
fermentation, Leuconostoc species increased again and Lactobacillus and Weissella decreased 364
at the end of the fermentation. These results suggest that there could be another important 365
determinant of microbial communities in addition to temperature and acidity. Data in Figs. 2 366
and 3 strongly indicate that the metabolism of free sugars and the production of lactate, 367
acetate, and mannitol closely correlated with the growth of Leuconostoc, Lactobacillus and 368
Weissella. 369
370
Comparative functional analysis of the kimchi microbiome. To explore the metabolic 371
potential of the kimchi microbiome during fermentation, all sequences were analyzed using 372
the SEED subsystem publicly available on the MG-RAST server. Assignment of orthologous 373
sequences in the metagenome by comparison with both protein and nucleotide databases were 374
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

16
categorized (28 metabolic categories, 519 subsystems). An average of 45.09% of total 375
sequences were matched to SEED functional categories of organisms present in the MG-376
RAST database at an E-value cutoff of 0.01 (Table 1). The level of reads categorized by 377
SEED were very low for the early fermentation, as only 3.33% and 7.45% of reads from J1 378
and J7 samples were categorized into the SEED functional categories, respectively. This 379
might be explained by a contamination of genomic DNA by cabbage and other kimchi 380
ingredients. As the kimchi fermentation progressed, matching levels of the metagenomic 381
sequence reads to SEED functional categories increased sharply due to the increase in 382
bacterial abundance (Fig. 1). 383
Results of assigning these metagenomic sequences to functional categories are shown in 384
Figs. 4 and 5. As expected, patterns in the data confirmed that kimchi fermentation is 385
accomplished predominantly by heterofermentative LAB, therefore carbohydrate categories 386
including metabolism of mono-, di-, and oligosaccharides and fermentation were key 387
categories for kimchi fermentation. Thus, among the many genes in the kimchi metagenome, 388
our analyses focused on carbohydrate categories. The comparative representation with several 389
other environmental metagenomic data sets showed that genes involved in the fermentation 390
process were enriched (Table 2). Carbohydrate metabolism yielded an average of 14.49% of 391
all matches (Fig. 4). The fraction of the kimchi microbiome reads in the carbohydrate 392
category was higher than those of microbiomes derived from the termite gut (12.89%), acid 393
mine drainage (AMD) biofilm (12.17%), enhanced biological phosphorus removal (EBPR) 394
sludge (10.72%), farm soil (11.25%) and Sargasso Sea (12.23%). Analysis of metabolic 395
potential within the carbohydrate category indicated that the kimchi microbiome was enriched 396
for genes involved and monosaccharide, di- and oligosaccharide fermentation. The 397
metagenomic read fractions of monosaccharide (28.70%), di- and oligosaccharide (28.38%), 398
and fermentation (14.38%) within the carbohydrate category in the kimchi microbiome were 399
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

17
also higher than in the microbiomes derived from the termite gut (24.35%, 21.64%, 5.24%), 400
AMD biofilm (7.12%, 24.1%, 10.51%), EBPR sludge (11.55%, 14.32%, 6.77%), farm soil 401
(18.41%, 15.3%, 7.52%) and Sargasso Sea (18.04%, 13.9%, 9.18%). The fermentation 402
metabolism subcategory was mostly associated with various lactate fermentations (65.93%) 403
and acetoin and butanediol metabolism (28.10%). In particular, lactate fermentation genes in 404
the kimchi microbiome were predominantly enriched compared to the termite gut (33.33%), 405
AMD biofilm (29.29%), EBPR sludge (27.19%), farm soil (25.66%), and Sargasso Sea 406
(16.07%). Moreover, metabolic genes involved in carbohydrate metabolism and fermentation 407
generally increased as the kimchi fermentation progressed (Figs. 4 and 5). In summary, the 408
above-mentioned results showed that the kimchi microbiome had high metabolic versatility 409
with respect to lactic acid fermentations, which were also supported by the profiles of free 410
sugars and fermentation products (Fig. 2). 411
412
Individual genome recruitment. Genome recruitment analysis is an insightful way to 413
examine the degree to which microorganisms in a given habitat are represented by completed 414
genomic sequences (17). Therefore, the metagenomic sequence reads were compared to all 415
complete and draft microbial genomes available from the NCBI 416
(http://www.ncbi.nlm.nih.gov/sites/genome) using the criterion of 95% nucleotide identity. 417
The two most highly recruited genomes were Leuconostoc mesenteroides and Lactobacillus 418
sakei, respectively (Fig. 6A). Prior reports have indicated that Le. mesenteroides is one of the 419
prominent species in kimchi fermentation (24, 34). However L. sakei has been better known 420
as a psychrotrophic LAB associated with fresh meat and fish (4). The reads that aligned with 421
the Le. mesenteroides subsp. mesenteroides ATCC 8293 and L. sakei subsp. sakei 23K 422
genomes often featured extremely high sequence identities (Figs. 6B and 6C), suggesting that 423
LAB closely related to these two reference strains were abundant in the fermented kimchi 424
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

18
samples. It seems clear that close relatives of these specific strains contribute to a large 425
fraction of the heterotrophic lactic acid fermentation in kimchi. Nevertheless, there were some 426
significant differences between the genomes of the two reference strains and the LAB 427
genomes of the kimchi samples, since certain reference sequences were not represented within 428
the metagenome sequence, implying that the kimchi habitat may feature specific selective 429
pressures and support diverse populations not yet isolated or sequenced. 430
Although Leuconostoc citreum KM20 was originally isolated from Chinese cabbage 431
kimchi (23), its degree of genomic match to the metagenome was relatively low (Fig. 6D). 432
Phylogenetic analysis based on 16S rRNA gene sequences showed that the genus Weissella 433
was one of the predominant bacterial groups during kimchi fermentation (Fig. 3). However, 434
the draft genome of W. paramesenteroides ATCC 33313 matched poorly to the metagenome 435
sequence (Fig. 6E). This was surprising and suggests that the metagenome sequences from 436
Weissella might be derived from other Weissella species such as W. koreensis S-5623T, 437
isolated from Chinese cabbage kimchi (27). 438
439
Assembly of pyrosequencing-derived sequencing reads and assignment of assembled 440
contigs. A very important aspect of metagenomic sequencing is the possibility of assembling 441
contigs derived from a single microorganism. Therefore, sequencing reads from the kimchi 442
metagenome, which comprised a total of 560,907 clean reads, were assembled resulting in a 443
total of 27,835 contigs. However, many sequences on the contigs were found to be split due to 444
frame shifts mostly caused by homopolymeric sequences, which are difficult to resolve by 445
454-pyrosequencing technology. The longest contig was 270,682 bps in size. One hundred 446
twenty seven contigs longer than 10 kb in size were classified according to their taxonomic 447
affiliations by BLASTN comparisons to the GenBank nonredundant nucleotide (nr/nt) 448
database and selection of the top BLASTN hits (see Table S1 in the supplemental material). 449
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

19
Most contigs were clearly derived from the predominant LAB, Leuconostoc mesenteroides 450
subsp. mesenteroides ATCC8293 and Lactobacillus sakei subsp. sakei 23K at the species level 451
(>97% sequence identity). Surprisingly, four contigs, 02778, 27827, 01645, and 03545 of 452
139,667 bp, 137,689 bp, 79,287 bp, and 10,233 bp, respectively, were classified as putative 453
bacteriophage fragments. Three of these contigs, 02778, 27827, and 01645 contained genes 454
for a putative bacteriophage major capsid protein (MCP), which is used frequently for phage 455
phylogenic analysis (3). Phylogenetic analysis based on the MCP sequences showed that 456
contigs, 02778, 27827, and 01645 were related to LAB phages (data not shown). Although 457
contig 03545 did not contain a gene for the putative bacteriophage MCP, most of genes in the 458
contig sequence were matched with the Leuconostoc phage 1-A4 complete genome (33) with 459
high identity (92%), indicating that contig 03545 was clearly able to be assigned as a 460
Leuconostoc phage (see Table S1 in the supplemental material). CRISPR elements are sites 461
containing multiple short palindromic repeats (23-47bp) that flank short “spacers” composed 462
of viral DNA (58); their presence in a bacterial genome represents a key evidence of 463
bacteriophage attack (58). Four CRISPR elements were detected from 127 contigs with longer 464
than 10 kb, suggesting that bacterial cells might be suffered from bacteriophage infection. 465
466
Relative abundance of the phage-related contigs. Bacteriophages are ubiquitous and 467
abundant components of microbial communities, and have significant influence upon bacterial 468
diversity, horizontal gene transfer, and genetic diversity in environmental habitats (43). By 469
counting the number of reads mapped to each phage-related contig and normalizing by the 470
total read count, it was possible to estimate the relative abundance of phages. Figure 7 shows 471
the frequency at which reads were observed for each phage-related contig in the kimchi 472
microbial samples. Putative phage sequences were detected early in the J13 sample (day 13), 473
and the numbers subsequently increased steadily throughout the fermentation. However, 474
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

20
putative phage sequences increased suddenly in the J25 sample, and their abundance 475
amounted to approximately 7% of the total reads. This large number of putative phage 476
sequences in the metagenomic data sets was unexpected because it was thought that most 477
phage (if present) escaped from the cell fraction obtained by centrifugation. Possible 478
explanations for phage detection include: the phages were replicating within the kimchi 479
microbiome and free phage particles adhered to either particulate matter or bacterial cells at 480
the time of harvesting (10, 17). 481
In any event, the high abundance of phage sequences in the cellular fraction from Kimchi 482
indicate that a remarkably high proportion of bacterial cells were suffering from infection and 483
perhaps from lytic phage attack. Although the frequency of putative phage genes was high at 484
the end of the fermentation, such levels are consistent with previously reported ranges of 485
phage-infected bacterial cells (10). It was inferred that the decrease in bacterial population 486
after day 25 (Fig. 1) and the major changes of microbial community between days 23 and 25 487
(Fig. 2) were affected by phage-host dynamics that occur during kimchi curing. With the 488
decrease of bacterial abundance after day 25, the putative phage sequences also slowly 489
decreased. Previous studies have reported that fermentation temperature and acidity are 490
important determinants of the microbial community composition in kimchi fermentation (6, 491
28). However, our results showed that in addition to temperature and acidity, a new influence, 492
bacteriophage was another clearly important determinant of microbial community dynamics 493
like sauerkraut fermentation (32, 33, 37), especially at the end stages of kimchi fermentation. 494
495
Conclusions. Changes in the overall features and metabolic potential of the kimchi-496
fermenting microbial community were investigated by examining a large pyrosequencing-497
derived data set, in conjunction with metabolite analysis. The metagenome and metabolites 498
revealed that the kimchi microbiome had high metabolic potential with respect to 499
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

21
heterotrophic lactic-acid fermentations. Based on the abundances of 16S rRNA genes and 500
representation of whole genomic sequences in the metagenome, we conclude that active 501
microbial populations in kimchi fermentation were dominated by members of three genera, 502
Leuconostoc, Lactobacillus, and Weissella, which are consistent with many previous results (6, 503
24, 28). Besides microbial genome sequences, a surprisingly high abundance of phage DNA 504
sequences were identified, indicating that bacteriophage influence the kimchi microbial 505
community and may be a key determinant of kimchi microbial community dynamics. 506
Metagenomic analysis of the kimchi samples over time made it possible to monitor not only 507
microbial community composition but also the overall features and metabolic potential during 508
fermentation. Future analysis of microbial abundances and interactions will likely provide 509
insights into how the kimchi microbial community influences metabolite production and, 510
ultimately, kimchi flavor. 511
512
ACKNOWLEDGMENTS 513
We thank professors J.S. Chun and C.J. Cha, and Drs. W.S. Park, B.H. Ryu, and Y.S. Ko for 514
their valuable help and collaboration during this study. This work was supported by the 515
Technology Development Program for Agriculture and Forestry (TDPAF) of the Ministry for 516
Agriculture, Forestry and Fisheries and the 21C Frontier Microbial Genomics and Application 517
Center Program (grant #: MG05-0104-4-0) of the Ministry of Education, Science and 518
Technology, Republic of Korea. 519
520
REFERENCES 521
1. Aziz, R. K., D. Bartels, A. A. Best, M. DeJongh, T. Disz, R. A. Edwards, K. Formsma, 522
S. Gerdes, E. M. Glass, M. Kubal, F. Meyer, G. J. Olsen, R. Olson, A. L. Osterman, R. A. 523
Overbeek, L. K. McNeil, D. Paarmann, T. Paczian, B. Parrello, G. D. Pusch, C. Reich, R. 524
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

22
Stevens, O. Vassieva, V. Vonstein, A. Wilke, and O. Zagnitko. 2008. The RAST Server: 525
rapid annotations using subsystems technology. BMC Genomics 9:75. 526
527
2. Bae, J. W., S. K. Rhee, J. R. Park, W. H. Chung, Y. D. Nam, I. Lee, H. Kim, and Y. H. 528
Park. 2005. Development and evaluation of genome-probing microarrays for monitoring 529
lactic acid bacteria. Appl. Environ. Microbiol. 71:8825-8835. 530
531
3. Bamford, D. H., J. M. Grimes, and D. I. Stuart. 2005. What does structure tell us about 532
virus evolution? Curr. Opin. Struct. Biol. 15:655-663. 533
534
4. Chaillou, S., M. C. Champomier-Vergès, M. Cornet, A. M. Crutz-Le Coq, A. M. Dudez, 535
V. Martin, S. Beaufils, E. Darbon-Rongère, V. Loux, R. Bossy, and M. Zagorec. 2005. 536
The complete genome sequence of the meat-borne lactic acid bacterium Lactobacillus sakei 537
23K. Nat. Biotechnol. 23:1527-1533. 538
539
5. Mcfeeters, R. F., and K. -H. Chen. 1986. Utilization of electron acceptors for anaerobic 540
mannitol metabolism by Lactobacillus plantarum. Compounds which serve as electron 541
acceptors. Food Science 3:73-81. 542
543
6. Cho, J., D. Lee, C. Yang, J. Jeon, J. Kim, and H. Han. 2006. Microbial population 544
dynamics of kimchi, a fermented cabbage product. FEMS Microbiol. Lett. 257:262-267. 545
546
7. Chou, H. H., and M. H. Holmes. 2001. DNA sequence quality trimming and vector 547
removal. Bioinformatics 17:1093-1104. 548
549
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

23
8. Cole, J. R., Q. Wang, E. Cardenas, J. Fish, B. Chai, R. J. Farris, A. S. Kulam-Syed-550
Mohideen, D. M. McGarrell, T. Marsh, G. M. Garrity, and J. M. Tiedje. 2009. The 551
ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic 552
Acids Res. 37:D141-D145. 553
554
9. Costello, E. K., C. L. Lauber, M. Hamady, N. Fierer, J. I. Gordon, and R. Knight. 555
2009. Bacterial community variation in human body habitats across space and time. Science 556
326:1694-1697. 557
558
10. DeLong, E. F., C. M. Preston, T. Mincer, V. Rich, S. J. Hallam, N. U. Frigaard, A. 559
Martinez, M. B. Sullivan, R. Edwards, B. R. Brito, S. W. Chisholm, and D. M. Karl. 2006. 560
Community genomics among stratified microbial assemblages in the ocean's interior. Science 561
311:496-503. 562
563
11. Dinsdale, E. A., R. A. Edwards, D. Hall, F. Angly, M. Breitbart, J. M. Brulc, M. 564
Furlan, C. Desnues, M. Haynes, L. L. Li, L. McDaniel, M. A. Moran, K. E. Nelson, C. 565
Nilsson, R. Olson, J. Paul, B. R. Brito, Y. J. Ruan, B. K. Swan, R. Stevens, D. L. 566
Valentine, R. V. Thurber, L. Wegley, B. A. White, and F. Rohwer. 2008. Functional 567
metagenomic profiling of nine biomes. Nature 452:629-632. 568
569
12. Ercolini, D. 2004. PCR-DGGE fingerprinting: novel strategies for detection of microbes 570
in food. J. Microbiol. Methods 56:297-314. 571
572
13. Edgar, R. C. 2004. MUSCLE: multiple sequence alignment with high accuracy and high 573
throughput. Nucleic Acids Res. 32:1792-1797. 574
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

24
575
14. Farelly, V., F. A. Rainley, and E. Stackebrandt. 1995. Effect of genome size and rrn 576
gene copy number on PCR amplification of 16S rRNA genes from a mixture of bacterial 577
species. Appl. Environ. Microbiol. 61:2798-2801. 578
579
15. Figueiredo, I. M., N. R. Pereira, P. Efraim, N. H. García, N. R. Rodrigues, A. 580
Marsaioli, Jr., and A. J. Marsaioli. 2006. 1H NMR, a rapid method to monitor organic acids 581
during cupuassu (Theobroma grandiflorum Spreng) processing. J. Agric. Food. Chem. 582
54:4102-4106. 583
584
16. García Martín, H., N. Ivanova, V. Kunin, F. Warnecke, K. W. Barry, A. C. 585
McHardy, C. Yeates, S. He, A. A. Salamov, E. Szeto, E. Dalin, N. H. Putnam, H. J. 586
Shapiro, J. L. Pangilinan, I. Rigoutsos, N. C. Kyrpides, L. L. Blackall, K. D. McMahon, 587
and P. Hugenholtz. 2006. Metagenomic analysis of two enhanced biological phosphorus 588
removal (EBPR) sludge communities. Nat. Biotechnol. 24:1263-1269. 589
590
17. Ghai, R., A. B. Martin-Cuadrado, A. G. Molto, I. G. Heredia, R. Cabrera, J. Martin, 591
M. Verdú, P. Deschamps, D. Moreira, P. López-García, A. Mira, and F. Rodriguez-592
Valera. 2010. Metagenome of the Mediterranean deep chlorophyll maximum studied by direct 593
and fosmid library 454 pyrosequencing. 4:1154-1166. 594
595
18. Gomez-Alvarez, V., T. K. Teal, and T. M. Schmidt. 2009. Systematic artifacts in 596
metagenomes from complex microbial communities. ISME J. 3:1314-1317. 597
598
19. Grissa, I., G. Vergnaud, and C. Pourcel. 2007. CRISPRFinder: a web tool to identify 599
clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35:W52-W57. 600
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

25
601
20. Ha, J. H., W. S. Hawer, Y. J. Kim, and Y. J. Nam. 1989. Changes of free sugars in 602
kimchi during fermentation. Korean J. Food Sci. Technol. 21:633-638 603
604
21. Juck, D., T. Charles, L. G. Whyte, and C. W. Greer. 2000. Polyphasic microbial 605
community analysis of petroleum hydrocarbon-contaminated soils from two northern 606
Canadian communities. FEMS Microbiol. Ecol. 33:241-249. 607
608
22. Kim, J., J. Chun, and H. U. Han. 2000. Leuconostoc kimchii sp. nov., a new species 609
from kimchi. Int. J. Syst. Evol. Microbiol. 50:1915-1919. 610
611
23. Kim, J. F., H. Jeong, J. S. Lee, S. H. Choi, M. Ha, C. G. Hur, J. S. Kim, S. Lee, H. S. 612
Park, Y. H. Park, and T. K. Oh. 2008. Complete Genome Sequence of Leuconostoc citreum 613
KM20. J. Bacteriol. 190:3093-3094. 614
615
24. Kim, M., and J. Chun. 2005. Bacterial community structure in kimchi, a Korean 616
fermented vegetable food, as revealed by 16S rRNA gene analysis. Int. J. Food Microbiol. 617
103:91-96. 618
619
25. Kim, N., K. R. Park, I. S. Park, Y. J. Cho, and Y. M. Bae. 2005. Application of a taste 620
evaluation system to the monitoring of Kimchi fermentation. Biosens. Bioelectron. 20:2283-621
2291. 622
623
26. Lee, J., C. Chun, M. Jung, J. Ahn, Y. Park, and T. Mheen. 1997. Classification of 624
isolates originating from kimchi using carbonsource utilization patterns. J. Microbiol. 625
Biotechnol. 7:68-74. 626
627
27. Lee, J., K. C. Lee, J. Ahn, T. Mheen, Y. Pyun, and Y. Park. 2002. Weissella koreensis 628
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

26
sp. nov., isolated from kimchi. Int. J. Syst. Evol. Microbiol. 52:1257-1261. 629
630
28. Lee, J. S., G. Y. Heo, W. L. Jun, Y. J. Oh, J. A. Park, Y. H. Park, Y. R. Pyun, and J. S. 631
Ahn. 2005. Analysis of kimchi microflora using denaturing gradient gel electrophoresis. Int. J. 632
Food Microbiol. 102:143-150. 633
634
29. Lee, D., S. Kim, J. Cho, and J. Kim. 2008. Microbial population dynamics and 635
temperature changes during fermentation of kimjang kimchi. J. Microbiol. 46:590-593. 636
637
30. Lee, E. J., R. Shaykhutdinov, A. M. Weljie, H. J. Vogel, P. J. Facchini, S. U. Park, Y. 638
K. Kim, and T. J. Yang. 2009. Quality assessment of ginseng by 1H NMR metabolite 639
fingerprinting and profiling analysis. J. Agric. Food Chem. 57:7513-7522. 640
641
31. Li, W. Z., and A. Godzik. 2006. Cd-hit: A fast program for clustering and comparing 642
large sets of protein or nucleotide sequences. Bioinformatics 22:1658-1659 643
644
32. Lu, Z., F. Breidt, V. Plengvidhya, and H. P. Flaming. 2003. Bacteriophage ecology in 645
commercial sauerkraut fermentations. Appl. Environ. Microbiol. 69:3192-3202. 646
647
33. Lu, Z., E. Altermann, F. Breidt, and S. Kozyavkin. 2010. Sequence analysis of 648
Leuconostoc mesenteroides bacteriophage ɸ1-A4 isolated from an industrial vegetable 649
fermentation. Appl. Environ. Microbiol. 76:1955-1966. 650
651
34. Makarova, K., A. Slesarev, Y. Wolf, A. Sorokin, B. Mirkin, E. Koonin, A. Pavlov, N. 652
Pavlova, V. Karamychev, N. Polouchin, V. Shakhova, I. Grigoriev, Y. Lou, D. Rohksar, 653
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

27
S. Lucas, K. Huang, D. M. Goodstein, T. Hawkins, V. Plengvidhya, D. Welker, J. 654
Hughes, Y. Goh, A. Benson, K. Baldwin, J.-H. Lee, I. Diaz-Muniz, B. Dosti, V. Smeianov, 655
W. Wechter, R. Barabote, G. Lorca, E. Altermann, R. Barrangou, B. Ganesan, Y. Xie, H. 656
Rawsthorne, D. Tamir, C. Parker, F. Breidt, J. Broadbent, R. Hutkins, D. O’Sullivan, J. 657
Steele, G. Unlu, M. Saier, T. Klaenhammer, P. Richardson, S. Kozyavkin, B. Weimer, 658
and D. Mills. 2006. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. 659
USA 103:15611-15616. 660
661
35. Meyer, F., D. Paarmann, M. D'Souza, R. Olson, E. M. Glass, M. Kubal, T. Paczian, 662
A. Rodriguez, R. Stevens, A. Wilke, J. Wilkening and R. A. Edwards. 2008. The 663
metagenomics RAST server - a public resource for the automatic phylogenetic and functional 664
analysis of metagenomes. BMC Bioinformatics 9:386-394. 665
666
36. Mou, X., S. L. Sun, R. A. Edwards, R. E. Hodson, and M. A. Moran. 2008. Bacterial 667
carbon processing by generalist species in the coastal ocean. Nature 451:708-711. 668
669
37. Mudgal, P., F., Jr. Breidt, S. R. Lubkin, and K. P. Sandeep. 2006. Quantifying the 670
significance of phage attack on starter cultures: a mechanistic model for population dynamics 671
of phage and their hosts isolated from fermenting sauerkraut. Appl. Environ. Microbiol. 672
72:3908-3915. 673
674
38. Nam, Y. D., H. W. Chang, K. H. Kim, S. W. Roh, and J. W. Bae. 2009. 675
Metatranscriptome analysis of lactic acid bacteria during kimchi fermentation with genome-676
probing microarrays. Int. J. Food Microbiol. 130:140-146. 677
678
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

28
39. Niu, B., L. Fu, S. Sun, and W. Li. 2010. Artificial and natural duplicates in 679
pyrosequencing reads of metagenomic data. BMC Bioinformatics 11:187-198. 680
681
40. Park, K. Y., E. J. Cho, S. H. Rhee, K. O. Jung, S. J. Yi, and B. H. Jhun. 2005. Kimchi 682
and an active component, beta-sitosterol, reduce oncogenic H-Ras(v12)-induced DNA 683
synthesis. J. Med. Food 6:151-156. 684
685
41. Park, E. J., H. W. Chang, K. H. Kim, Y. D. Nam, S. W. Roh, and J. W. Bae. 2009. 686
Application of quantitative real-time PCR for enumeration of total bacterial, archaeal, and 687
yeast populations in kimchi. J. Microbiol. 47:682-685. 688
689
42. Park, J. M., J. H. Shin, D. W. Lee, J. C. Song, H. J. Suh, U. J. Chang, and J. M. Kim. 690
2010. Identification of the lactic acid bacteria in kimchi according to initial and over-ripened 691
fermentation using PCR and 16S rRNA gene sequence analysis. Food Sci. Biotechnol. 692
19:541-546. 693
694
43. Parsley, L. C., E. J. Consuegra, S. J. Thomas, J. Bhavsar, A. M. Land, N. N. Bhuiyan, 695
M. A. Mazher, R. J. Waters, K. E. Wommack, W. F. Harper, Jr., and M. R. Liles. 2010. 696
Census of the viral metagenome within an activated sludge microbial assemblage. Appl. 697
Environ. Microbiol. 76:2673-2677. 698
699
44. Parsley, L. C, E. J. Consuegra, K. S. Kakirde, A. M. Land, W. F. Harper, Jr., M. R. 700
Liles. 2010. Identification of diverse antimicrobial resistance determinants carried on 701
bacterial, plasmid, or viral metagenomes from an activated sludge microbial assemblage. 702
Appl. Environ. Microbiol. 76:3753-3757. 703
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

29
704
45. Qin, J., R. Li, J. Raes, M. Arumugam, K. S. Burgdorf, C. Manichanh, T. Nielsen, N. 705
Pons, F. Levenez, T. Yamada, D. R. Mende, J. Li, J. Xu, S. Li, D. Li, J. Cao, B. Wang, H. 706
Liang, H. Zheng, Y. Xie, J. Tap, P. Lepage, M. Bertalan, J. M. Batto, T. Hansen, D. Le 707
Paslier, A. Linneberg, H. B. Nielsen, E. Pelletier, P. Renault, T. Sicheritz-Ponten, K. 708
Turner, H. Zhu, C. Yu, S. Li, M. Jian, Y. Zhou, Y. Li, X. Zhang, S. Li, N. Qin, H. Yang, 709
J. Wang, S. Brunak, J. Dore, F. Guarner, K. Kristiansen, O. Pedersen, J. Parkhill, J. 710
Weissenbach, P. Bork, S. D. Ehrlich, and J. Wang. 2010. A human gut microbial gene 711
catalogue established by metagenomic sequencing. Nature 464:59-65. 712
713
46. Ritalahti, K. M., B. K. Amos, Y. Sung, Q. Wu, S. S. Koenigsberg, and F. E. L�ffler. 714
2006. Quantitative PCR targeting 16S rRNA and reductive dehalogenase genes 715
simultaneously monitors multiple Dehalococcoides strains. Appl. Environ. Microbiol. 716
72:2765-2774. 717
718
47. Rodriguez-Brito, B., L. Li, L. Wegley, M. Furlan, F. Angly, M. Breitbart, J. 719
Buchanan, C. Desnues, E. Dinsdale, R. Edwards, B. Felts, M. Haynes, H. Liu, D. Lipson, 720
J. Mahaffy, A. B. Martin-Cuadrado, A. Mira, J. Nulton, L. Pašic, S. Rayhawk, J. 721
Rodriguez-Mueller, F. Rodriguez-Valera, P. Salamon, S. Srinagesh, T. F. Thingstad, T. 722
Tran, R. V. Thurber, D. Willner, M. Youle, and F. Rohwer. 2010. Viral and microbial 723
community dynamics in four aquatic environments. ISME J. 4:739-751. 724
725
48. Roh, S. W., K. H. Kim, Y. D. Nam, H. W. Chang, E. J. Park, and J. W. Bae. 2010. 726
Investigation of archaeal and bacterial diversity in fermented seafood using barcoded 727
pyrosequencing. ISME J. 3:745-751. 728
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

30
729
49. Sanapareddy, N., T. J. Hamp, L. C. Gonzalez, H. A. Hilger, A. A. Fodor, and S. M. 730
Clinton. 2009. Molecular diversity of a North Carolina wastewater treatment plant as 731
revealed by pyrosequencing. Appl. Environ. Microbiol. 75:1688-1696. 732
733
50. Shendure. J., and H. Ji. 2008. Next-generation DNA sequencing. Nat. Biotechnol. 734
26:1135-1145. 735
736
51. Shin, M. S., S. K. Han, J. S. Ryu, K. S. Kim, and W. K. Lee. 2008. Isolation and partial 737
characterization of a bacteriocin produced by Pediococcus pentosaceus K23-2 isolated from 738
kimchi. J. Appl. Microbiol. 105:331-339. 739
740
52. Simon, C., A. Wiezer, A. W. Strittmatter, and R. Daniel. 2009. Phylogenetic diversity 741
and metabolic potential revealed in a glacier ice metagenome. Appl. Environ. Microbiol. 742
75:7519-7526. 743
744
53. Son, H. S., K. M. Kim, F. van den Berg, G. S. Hwang, W. M. Park, C. H. Lee, and Y. 745
S. Hong. 2008. 1H nuclear magnetic resonance-based metabolomic characterization of wines 746
by grape varieties and production areas. J. Agric. Food Chem. 56:8007-8016. 747
748
54. Song, Y. O. 2004. The functional properties of kimchi for the health benefits. J. Food Sci. 749
Nutri. 9:27-33. 750
751
55. Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: molecular evolutionary 752
genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596-1599. 753
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

31
754
56. Tringe, S. G., C. von Mering, A. Kobayashi, A. A. Salamov, K. Chen, H. W. Chang, M. 755
Podar, J. M. Short, E. J. Mathur, J. C. Detter, P. Bork, P. Hugenholtz, and E. M. Rubin. 756
2005. Comparative metagenomics of microbial communities. Science 308:554-557. 757
758
57. Tyson, G. W., J. Chapman, P. Hugenholtz, E. E. Allen, R. J. Ram, P. M. Richardson, 759
V. V. Solovyev, E. M. Rubin, D. S. Rokhsar, and J. F. Banfield. 2004. Community 760
structure and metabolism through reconstruction of microbial genomes from the environment. 761
Nature 428:37-43. 762
763
58. Tyson, G. W., and J. F. Banfield. 2008. Rapidly evolving CRISPRs implicated in 764
acquired resistance of microorganisms to viruses. Environ. Microbiol. 10:200-207. 765
766
59. Venter, J. C., K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. 767
Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, 768
K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. 769
Pfannkoch, Y.-H. Rogers, and H. O. Smith. 2004. Environmental genome shotgun 770
sequencing of the Sargasso Sea. Science 304:66-74. 771
772
60. Wilmes, P., S. L. Simmons, V. J. Denef, and J. F. Banfield. 2009. The dynamic genetic 773
repertoire of microbial communities. FEMS Microbiol. Rev. 33:109-132. 774
775
61. Wisselink, H. W., R. A. Weusthuis, G. Eggink, J. Hugenholtz, and G. J. Grobben. 776
2002. Mannitol production by lactic acid bacteria: a review. Int. Dairy J. 12:151-161. 777
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

32
778
FIGURE LEGENDS 779
FIG. 1. Change in pH and 16S rRNA gene copy number for total bacterial numbers during 780
kimchi fermentation. Measurements of 16S rRNA gene copy numbers were performed 781
independently in triplicate. Error bars represent the standard deviation. 782
783
FIG. 2. Quantification of identified key metabolites in kimchi supernatants during 784
fermentation by 1H-NMR. Data are given as means ± standard error, calculated from three 785
replicates. Quantification was determined using the Chenomx NMR Suite v. 6.1 with 2,2-786
dimethyl-2-silapentane-5-sulfonate (DSS) as the internal standard. 787
788
FIG. 3. Phylogenetic taxonomic composition of the kimchi microbiome during fermentation. 789
16S rRNA gene sequences were classified to the genus level using the MG-RAST sever based 790
on the RDP (16S rRNA gene) database (E-value, 0.01; minimum alignment length, ≥50 bp). 791
792
FIG. 4. Predicted metabolic profiles of kimchi metagenomic data sets obtained from SEED 793
subsystems. The MG-RAST server based on SEED subsystems provided categories for the 794
metagenomic data sets. The MG-RAST recommended parameters (E-value, 0.01; minimum 795
sequence length, ≥50 bp) were used. 796
797
FIG. 5. Predicted metabolic profiles of kimchi metagenomic data sets related to carbohydrate 798
metabolism and fermentation. Categories for the metagenomic data sets were provided by the 799
MG-RAST server based on SEED subsystems. The MG-RAST recommended parameters (E-800
value, 0.01; minimum sequence length, ≥50 bp) were used. 801
802
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

33
FIG. 6. Recruitment of whole clean reads against complete or draft genomes (A). Insets 803
are the recruitment plots for the four top highest recruiting genomes; Leuconostoc 804
mesenteroides subsp. mesenteroides ATCC 8293, (B); Lactobacillus sakei subsp. sakei 23805
K, (C); Leuconostoc citreum KM20, (D); and Weissella paramesenteroides ATCC 33313, 806
(E). Lines within the recruitment plots indicate the level of 95% nucleotide identity. 807
808
FIG. 7. Relative abundance of putative phage contigs in whole unassembled clean reads 809
during kimchi fermentation. Kimchi metagenomic data sets were matched against four 810
putative phage contigs using the BLASTN algorithm with the criteria of a minimum percent 811
identity of 95% and a minimum overlap of 90 bp. 812
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

34
813
TABLE 1. Summary of the pyrosequencing-derived data sets, and statistical analysis of, 814
kimchi fermentation samples. 815
Kimchi samples J1 J7 J13 J16 J18 J21 J23 J25 J27 J29 Total
Total size of sequences (bp) 11417220 15705531 9806465 13411871 8009837 27622094 28190450 104312483 49419903 25183023 281661657
Number of reads 27337 38388 23287 31647 18846 66096 67771 249849 118313 60022 701556
Size of clean sequences (bp) 9646351 13090858 8301534 11390298 6765854 23238193 23565573 86509803 41595359 21345546 245449369
Number of clean reads 22006 30202 18885 25737 15208 53106 53919 197709 95322 48813 560907
Average length of clean reads (bp) 438 433 439 442 444 437 437 437 436 437 438
Average GC (%) of clean reads§ 37.53 37.25 37.54 38.62 38.03 37.38 37.49 38.13 37.81 37.26 37.70
Number (%†) of
predicted 16S rRNA genes*
38
(0.17)
57
(0.19)
90
(0.48)
199
(0.77)
101
(0.66)
300
(0.56)
334
(0.62)
1316
(0.67)
592
(0.62)
252
(0.52)
3279
(0.59)
Number (%†) of reads categorized
by SEED subsystem*
732
(3.33)
2249
(7.45)
8059
(42.67)
12419
(48.25)
8865
(58.29)
23849
(44.91)
24486
(46.11)
102770
(51.98)
43808
(45.96)
23406
(47.95)
252936
(45.09)
§Fractional GC contents of DNA sequence was calculated by GEECEE program (http://emboss.bioinformatics.nl/cgi-816
bin/emboss/geecee). 817
†The numbers in parentheses indicate the percentages of respective read numbers for clean read numbers. 818
*An E-value of less than 0.01 and a minimum alignment length (≥ 50 bp) was used to predict 16S rRNA and functional genes 819
using the MG-RAST server. 820 on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

35
821
TABLE 2. Comparative representation of the major metabolic groups of the kimchi 822
microbiome versus several other environmental metagenomic data sets publicly available in 823
MG-RAST 824
Relative abundance (%)*
SEED class SEED category Kimchi
Termite gut
(4442701.3)§
AMD biofilm
(4441137.3)§
Farm soil
(4441091.3)§
EBPR sludge
(4441092.3)§
Sargasso Sea
(4441571.3)§
Subsystem
hierarchy 1 Carbohydrate 14.49 12.89 12.17 11.25 10.72 12.23
Uptake system 0.07 0.64 0.11 1.80 0.51 0.51
Di-and oligosaccharides 28.38 21.64 24.10 15.30 14.33 13.90
Central carbohydrate metabolism 18.05 26.16 39.86 26.67 30.47 28.63
Amino sugars 1.66 7.90 1.88 4.71 2.65 2.47
One-carbon metabolism 0.69 2.60 3.28 8.15 11.09 8.76
Organic acids 2.82 3.22 6.12 5.34 8.33 8.10
Quorum sensing and biofilm formation 0.35 0.55 0.16 0.40 0.61 0.54
Fermentation 14.38 5.24 10.51 7.52 6.77 9.18
CO2 fixation 0.74 2.17 3.60 4.46 7.78 2.33
Sugar alcohols 4.17 4.98 3.24 6.96 5.68 7.35
Methanogenesis - 0.43 0.02 0.22 0.22 0.10
Monosaccharides 28.70 24.35 7.12 18.41 11.55 18.04
Subsystem
hierarchy 2
Carbohydrate - 0.13 - 0.07 0.01 0.09
Butanol biosynthesis 2.60 10.16 3.57 20.21 21.20 21.27
Fermentation (mixed acid) 2.72 8.54 5.79 3.54 7.92 3.90
Fermentation (lactate) 65.93 33.33 29.29 25.66 27.19 16.08
Acetyl-CoA fermentation to butylate 0.65 18.29 43.71 30.97 32.33 42.23
Subsystem
Acetoin, butanediol metabolism 28.10 29.67 17.64 19.62 11.35 16.54
* Relative abundance means the relative proportion of sequencing reads performing specific metabolisms within each subsystem. 825 § Project identifying numbers in the MG-RAST system. 826
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

36
827
Figure 1 828
829
830
831
Days
0 5 10 15 20 25 30
16
S r
RN
A g
ene
cop
ies
/ml
108
109
1010
1011
pH
4.2
4.6
5.0
5.4
5.816S rRNA gene copies
pH
832
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from
37
833
Figure 2 834
835
836
837
838
Days
0 5 10 15 20 25 30
Co
nce
ntr
ati
on
(m
M)
0
20
40
60
80Glucose
Fructose
Sucrose
Mannitol
Lactate
Acetate
Ethanol
839
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

38
840
Figure 3 841
842
843
844
845
846
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

39
847
Figure 4 848
849
850
851
852
853
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

40
854
Figure 5 855
856
857
858
859
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

41
860
Figure 6 861
862
863
864
865
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from

42
866
Figure 7 867
868
869
870
Kimchi samples
J1 J7 J13 J16 J18 J21 J23 J25 J27 J29
Rel
ati
ve
ab
un
dan
ce (
%)
0
1
2
3
4
5
6
7
8
contig 03545
contig 01645
contig 27827
contig 02778
871
872
873
on May 16, 2019 by guest
http://aem.asm
.org/D
ownloaded from