nuclease-deficient fen-1 blocks rad51/brca1-mediated repair and causes trinucleotide
TRANSCRIPT

MOLECULAR AND CELLULAR BIOLOGY, Sept. 2003, p. 6063–6074 Vol. 23, No. 170270-7306/03/$08.00�0 DOI: 10.1128/MCB.23.17.6063–6074.2003Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Nuclease-Deficient FEN-1 Blocks Rad51/BRCA1-Mediated Repair andCauses Trinucleotide Repeat Instability
Craig Spiro1 and Cynthia T. McMurray1,2*Department of Molecular Pharmacology and Experimental Therapeutics1 and Department of Biochemistry and Molecular Biology
and Molecular Neuroscience Program,2 Mayo Clinic and Foundation, Rochester, Minnesota 55905
Received 13 March 2003/Returned for modification 22 April 2003/Accepted 8 May 2003
Previous studies have shown that expansion-prone repeats form structures that inhibit human flap endo-nuclease (FEN-1). We report here that faulty processing by FEN-1 initiates repeat instability in mammaliancells. Disease-length CAG tracts in Huntington’s disease mice heterozygous for FEN-1 display a tendencytoward expansions over contractions during intergenerational inheritance compared to those in homozygouswild-type mice. Further, with regard to human cells expressing a nuclease-defective FEN-1, we provide directevidence that an unprocessed FEN-1 substrate is a precursor to instability. In cells with no endogenous defectsin DNA repair, exogenous nuclease-defective FEN-1 causes repeat instability and aberrant DNA repair.Inefficient flap processing blocks the formation of Rad51/BRCA1 complexes but invokes repair by otherpathways.
Trinucleotide repeats give rise to disease when the numberof repeated units of a simple trinucleotide, such as CAG,expands beyond an unaffected range (43). In the unaffectedrange, the number of repeated trinucleotide units, thoughpolymorphic, is stable. An increase in the number of repeatedunits beyond a certain threshold, however, causes instability aswell as disease, and expansion in subsequent generations be-comes likely (21, 43). While the molecular events that give riseto repeat instability remain obscure, studies during the lastseveral years have given sufficient insight to allow the devel-opment of working models for expansion. Emerging data sug-gest that expansion arises in the process of break repair (15, 35,50, 51) and is mediated by aberrant interaction of key DNArepair enzymes. Important among these is the flap endonucle-ase (FEN-1).
FEN-1 is a structure-specific nuclease that participates inDNA replication and repair (39). Flap endonuclease (humanFEN-1 and/or Saccharomyces cerevisiae Rad27 protein) is re-quired for normal maturation of Okazaki fragments duringreplication (31, 39, 49, 68). FEN-1 has also been implicated inseveral repair processes, including base excision repair (BER)(13, 32, 33, 47, 49, 56, 64), nonhomologous end joining (70),recombination (46), nucleotide excision repair (NER) (57),and removal of bulky UV lesions by an alternative excisionpathway (72). The details of the steps by which flap endonu-clease protects cells from repeat instability or causes expansionremain unclear. However, yeast lacking Rad27 shows both anincreased rate of expansion of trinucleotide repeats (15, 52, 60)and duplications flanked by direct repeats (29, 62, 71). Thesedata have been used to support the notion that defective flapprocessing plays an important causative role in expansion. Thefailure of FEN-1 to process flaps folded into aberrant hairpinstructures is thought to cause expansion at CAG repeats (26,
60). FEN-1 is inhibited by hairpins comprising CAG triplets,because the 5� end needed for FEN-1 loading is concealedwithin the hairpin structure (26, 60).
Despite the compelling data that loss of yeast Rad27 causesexpansion, the relevance to human instability is uncertain. Thehuman FEN-1 and yeast Rad27 protein are homologous, andthe human enzyme can complement the defect of yeast null forRad27 protein (22, 23). Yet the features of expansion inducedby loss of Rad27 in yeast are very different from those observedin human disease.
In yeast, expansions are rare compared to deletions, while inhuman diseases, expansions exceed contractions (16, 42–44,52). Deletion of Rad27 from yeast causes a severe growthdefect at 37°C, instability throughout the genome (mutatorphenotype), and increased recombination (30, 45, 61, 62). Inmammals, however, loss of FEN-1 is embryonically lethal (37).In human expansion disease, there is neither exchange noralterations of flanking sequence around the CAG expandedrepeat region, suggesting that expansion is not associated withincreased homologous recombination. Further, in human re-peat expansion, the mutation is limited to a single site (20),inconsistent with a mutator phenotype as occurs with the lossof flap endonuclease in yeast (30, 51). Expansion in humandisease must, therefore, occur in the presence of a normalFEN-1 protein. The majority of expansion studies of yeast haverelied on models from which the flap endonuclease (Rad27gene product) is absent. Taken together, the features of rad27null mutants in yeast do not recapitulate many of the featuresof expansion in mammalian disease. This raises a question asto the role of FEN-1 in expansion in mammals.
We report here studies designed to improve understandingof the role of FEN-1 in repeat instability in mammals. We havecrossed a transgenic mouse lacking one functional allele forFEN-1 (37) with transgenic animals harboring an expandedCAG repeat within the human Huntington’s disease (hHD)gene (40). Mice heterozygous for FEN-1 develop and repro-duce normally, but they have a rapid tumor progression phe-
* Corresponding author. Mailing address: Mayo Clinic, 200 First St.SW, Rochester, MN 55905. Phone: (507) 284-1597. Fax: (507) 284-9111. E-mail: [email protected].
6063
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

notype (37). Experiments using these animals, therefore, havethe advantage that expansion in the hHD allele can be evalu-ated with a reduced level of normal FEN-1 rather than in itsabsence. Additionally, we evaluated trinucleotide instability ofendogenous repeats in human cells stably transformed with anuclease-defective FEN-1 that retains its ability to supportprotein-protein interactions.
FEN-1 protects mammalian cells from repeat instability. Ex-pression of nuclease-defective enzyme destabilizes microsatel-lites. In addition, cells expressing nuclease-defective FEN-1show aberrant foci containing proteins of break repair andNER pathways.
MATERIALS AND METHODS
Transgenic and heterozygous knockout mice. The transgenic mouse R6/1 hasexon 1 of an hHD allele containing about 120 CAG repeats, as described byMangiarini et al. (40). Heterozygous knockout FEN-1 mice were from R.Kucherlapati (Harvard Medical School) (37); FEN-1 mice contain one wild-type(wt) and one nonfunctional flap endonuclease allele (37). To produce progenythat contained one hHD allele and were heterozygous for FEN-1, male R6/1mice were bred with female heterozygous FEN-1 mice as described previously(36). A duplex PCR was used to determine the FEN-1 genotype. Primer A (5�GGGAGTGAGATGGCAGTGTT, corresponding to a sequence within theFEN-1 coding region, nucleotides [nt] 5539 to 5558 in GenBank AY014962) andprimer C (5� GGCACTCAGGGTGTTTTCAA, corresponding to a sequence inthe 3� flanking region, nt 5832 to 5813) give a 293-bp product from the wt allele;primer B (5� TGGAAGGATRTGGAGCTACGGC, which corresponds to asequence within the targeting vector) and primer C give a 373-bp product fromthe interrupted allele. The presence of the hHD allele and the length of the allelewere determined using primers described previously (40). GraphPad Prism wasused for statistical calculations.
Plasmids, cell cultures, and transformation. HCC1937, a BRCA1 mutatedbreast cancer cell line (65), and the HCC1937 � 5� mycBRCA1-revertant line (7,53, 65) were from J. Chen (Mayo Clinic). HCC1937 and SK-N-MC cells (Amer-ican Type Culture Collection) were maintained in RPMI 1640–10% fetal calfserum. wt FEN-1 cDNA with a hemagglutinin [HA] epitope tag encoded at the3� end and D181A mutant FEN-1 cDNA were gifts of M. S. Park (Los AlamosNational Laboratory). The coding region for nuclease-deficient FEN-1 D181Acontains a C-for-A substitution at nt 542, which changes codon 181 from GAC(aspartate) in the wt to GCC (alanine) (54–56) (see Fig. 2.) A 625-bp Bpu1102Ifragment (nt 433 to 1058) within the FEN-1 D181A coding region was substi-tuted for the Bpu1102I fragment in the wt FEN-1-HA cDNA to create theD181A-HA cDNA. The wt and D181A coding regions were inserted into thevector pcDNA3.1/Hygro (Invitrogen), and plasmids were sequenced to confirmthat they differed at the single nucleotide only. Cells were transformed by plas-mid DNA-Lipofectamine (Invitrogen). At 24 h after transformation, growthmedium was replaced by growth medium supplemented with 100 to 150 �g ofhygromycin/ml, a concentration toxic to the SK-N-MC cells. Colonies surviving ina single well were pooled. Using Lipofectamine 2000 (Invitrogen) for transientassays, SK-N-MC cells were cotransformed with the appropriate FEN-1 con-struct and pEGFP-N1, a green fluorescent protein (GFP) expression vector(Clontech).
Assay for FEN-1 protein. Expression of exogenous FEN-1 was determined byimmunoblotting using a rabbit anti-HA antibody (Zymed). Cells were harvestedinto sodium dodecyl sulfate sample buffer, cellular proteins were resolved by12% polyacrylamide gel electrophoresis, and proteins were transferred to nitro-cellulose by semidry blotting as described previously (58). After incubatingmembranes with anti-HA, HA-containing proteins were visualized using goatanti-rabbit horseradish peroxidase-conjugated antibody and West Pico chemilu-minescent substrate (Pierce). The calculated molecular mass of the FEN wt-HAfusion protein was 44,164 Da.
FEN-1 nuclease activity. For analysis of FEN-1 activity, nuclear extract fromFEN-1-transformed and parent SK-N-MC cells was prepared using the methodof Andrews and Faller (2), diluted to a final NaCl concentration of 200 mM, andassayed (using the modified Bradford method [Bio-Rad]) for total protein.Flap-1 substrate was used to measure FEN-1 activity in a modification of thepreviously described assay (24, 60). The reaction mixture included 50 mM Tris-Cl(pH 8), 10 mM MgCl2, 0.5 mM �-mercaptoethanol, 100 �g of bovine serumalbumin (BSA)/ml, 33 �g of sonicated salmon sperm DNA/ml, 80 fmol of 5�-end
32P-labeled substrate, and nuclear extract. The substrate and cleaved productwere resolved on 6% sequencing gels and analyzed by PhosphorImager (Molec-ular Dynamics).
Microsatellite instability assays. SK-N-MC and FEN-transformed cells wereharvested, and DNA was purified as described previously (59). Repetitive alleleswere amplified using conditions that have been previously described and con-firmed by independently repeating the assay (18, 20). For GeneScan (version 3.0;PE Applied Biosystems) analysis, PCR was carried out with a single fluorescein-labeled primer and the products were resolved by polyacrylamide gel electro-phoresis. The products were identified by scans of the gel, so that peaks corre-sponded to DNA molecules. The prominent peak determines the repeat lengthof the allele. Using values from a GeneScan table, a fraction in a prominent bandwas calculated for each allele to compare samples. For example, the fractionunder peak 191 equals the value for 191 divided by the sum of the values for 191and 188 [see Fig. 3]). For the ACTC and HD alleles, instability was tested byamplification with alternative sets of primers. For ACTC, the first primer set was51 to 70 with 140 to 121 (20); the second set was 51 to 70 with 152 to 133(numbering according to GenBank accession no. HSAC06). For the HD allele,the first primer set was 337 to 366 with 485 to 465 (40); the second set was 308to 332 with 485 to 465 (numbered according to GenBank accession no. HUM-HDA).
Drug treatment and cell survival. Approximately 300 to 600 cells were platedin series either in the wells of 6-well dishes or in 60-mm-diameter dishes. Cellswere allowed to attach overnight. Methyl methanesulfonate (MMS) and cisplatinwere diluted into phosphate-buffered saline (PBS), and the stocks were sterilizedby filtration through a 0.2-�m-pore-size filter. Drugs were added to cultures fromthe concentrated stock. After a 1-h exposure to drugs, the medium was aspiratedand replaced with complete growth medium lacking drugs. At 10 to 12 days afterdrug treatment, cells were fixed and stained in a solution of 2% methylene bluein 50% methanol. Colonies were counted, and the fraction of surviving cells wasdetermined (colonies in treated dish divided by colonies in zero-drug control forthat series). Data are shown for series that used the same drug dilutions. Data forcisplatin represent averages for six series. For MMS, data are averages for threeseries.
Immunofluorescence assay of nuclear foci. Anti-FEN-1 (N-17), anti-ERCC1(FL-297), anti-XPG (N-17), anti-p21 (C-19), anti-CBP (A-22), and anti-Rad51(H-92) were from Santa Cruz Biotechnology. Anti-rabbit Cy5 was from Amer-sham. Anti-goat Cy5 was from Zymed. Cells were plated on coverslips in thewells of 6-well dishes 1 day before drug treatment. Cisplatin stock (2 mM) wasprepared in PBS and added (15 �M final concentration) to cultures at theindicated time before time zero. At time zero, coverslips were washed with PBS,fixed in ice-cold methanol, washed, and then incubated sequentially in blockingbuffer (Tris-buffered saline [TBS] containing 0.02% Tween 20 [Sigma], 5% skimmilk [Difco], 5% BSA [Roche]) and in primary antibody diluted in blockingbuffer. Cells were washed in TBS–0.02% Tween 20 and then incubated in sec-ondary antibody conjugated to Cy5. The integrity of compartments and primaryantibody-dependent staining was confirmed (see Results and Fig. 5). As a controlfor primary antibody specificity, mock experiments in which a primary antibodyraised in a different species was used with the same secondary antibody werecarried out in parallel (see Fig. 5). Coverslips were then washed in TBS–0.02%Tween 20, in TBS, and in water containing 0.2 �g of Hoechst 33258/ml to stainnuclei. To estimate the number of nuclear foci, coverslips were examined for Cy5fluorescent dots within the area of Hoechst staining. Foci in 50 unselected nucleiwere counted. The average number is reported (� the standard error of themean [SEM]).
RESULTS
Instability at a disease-length allele in mice heterozygousfor FEN-1. The R6/1 mouse contains exon 1 of the hHD genewith an expanded CAG repeat (about 120 CAG [40]). Therepeat is unstable during intergenerational inheritance and insomatic tissue (in an age-dependent manner) (35, 40, 41). Totest the hypothesis that FEN-1 contributes to trinucleotideexpansion in mammals, we crossed the FEN-1�/� mice (37)with R6/1 mice (Fig. 1A) and evaluated the behavior of theexpanded CAG trinucleotide tract in the FEN-1�/�/hHD�/�
progeny (Fig. 1B; Table 1).DNA from tails of 3-week-old offspring was analyzed for
CAG repeat length at the hHD allele (36). The repeat length
6064 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.
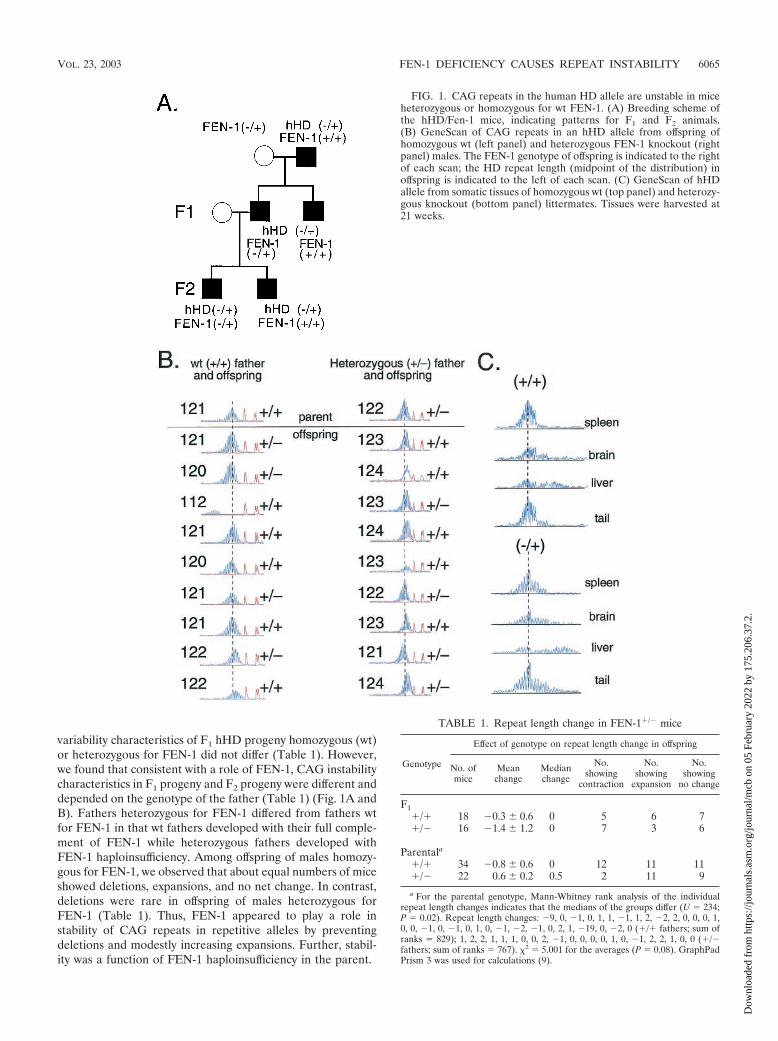
variability characteristics of F1 hHD progeny homozygous (wt)or heterozygous for FEN-1 did not differ (Table 1). However,we found that consistent with a role of FEN-1, CAG instabilitycharacteristics in F1 progeny and F2 progeny were different anddepended on the genotype of the father (Table 1) (Fig. 1A andB). Fathers heterozygous for FEN-1 differed from fathers wtfor FEN-1 in that wt fathers developed with their full comple-ment of FEN-1 while heterozygous fathers developed withFEN-1 haploinsufficiency. Among offspring of males homozy-gous for FEN-1, we observed that about equal numbers of miceshowed deletions, expansions, and no net change. In contrast,deletions were rare in offspring of males heterozygous forFEN-1 (Table 1). Thus, FEN-1 appeared to play a role instability of CAG repeats in repetitive alleles by preventingdeletions and modestly increasing expansions. Further, stabil-ity was a function of FEN-1 haploinsufficiency in the parent.
FIG. 1. CAG repeats in the human HD allele are unstable in miceheterozygous or homozygous for wt FEN-1. (A) Breeding scheme ofthe hHD/Fen-1 mice, indicating patterns for F1 and F2 animals.(B) GeneScan of CAG repeats in an hHD allele from offspring ofhomozygous wt (left panel) and heterozygous FEN-1 knockout (rightpanel) males. The FEN-1 genotype of offspring is indicated to the rightof each scan; the HD repeat length (midpoint of the distribution) inoffspring is indicated to the left of each scan. (C) GeneScan of hHDallele from somatic tissues of homozygous wt (top panel) and heterozy-gous knockout (bottom panel) littermates. Tissues were harvested at21 weeks.
TABLE 1. Repeat length change in FEN-1�/� mice
Genotype
Effect of genotype on repeat length change in offspring
No. ofmice
Meanchange
Medianchange
No.showing
contraction
No.showing
expansion
No.showing
no change
F1�/� 18 �0.3 � 0.6 0 5 6 7�/� 16 �1.4 � 1.2 0 7 3 6
Parentala
�/� 34 �0.8 � 0.6 0 12 11 11�/� 22 0.6 � 0.2 0.5 2 11 9
a For the parental genotype, Mann-Whitney rank analysis of the individualrepeat length changes indicates that the medians of the groups differ (U � 234;P � 0.02). Repeat length changes: �9, 0, �1, 0, 1, 1, �1, 1, 2, �2, 2, 0, 0, 0, 1,0, 0, �1, 0, �1, 0, 1, 0, �1, �2, �1, 0, 2, 1, �19, 0, �2, 0 (�/� fathers; sum ofranks � 829); 1, 2, 2, 1, 1, 1, 0, 0, 2, �1, 0, 0, 0, 0, 1, 0, �1, 2, 2, 1, 0, 0 (�/�fathers; sum of ranks � 767). �2 � 5.001 for the averages (P � 0.08). GraphPadPrism 3 was used for calculations (9).
VOL. 23, 2003 FEN-1 DEFICIENCY CAUSES REPEAT INSTABILITY 6065
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

Previous studies with R6/1 mice have revealed that repeatsappear to be stable in somatic tissues until 11 weeks but thatexpansion occurs with aging (35, 40, 41). The degree of age-dependent expansion varies with the tissue type (35, 40, 41).We therefore examined tissues of littermates at 21 weeks ofage to determine whether the degree of somatic instabilitydiffers depending on FEN-1 genotype. However, we found nosignificant differences in CAG repeat length in the hHD alleleamong tissues from FEN-1�/�/hHD�/� and FEN-1�/�/hHD�/� littermates sacrificed at different ages. Therefore,CAG repeats in somatic cells did not show FEN-1 genotype-dependent changes (Fig. 1C). FEN-1 effects on instability inprogeny were limited to the developing germ cells.
Analyses of mice, then, indicate that haploinsufficiency ofFEN-1 can alter trinucleotide repeat length stability by inhib-iting the frequency of deletion events and modestly increasingexpansion events at long disease-length alleles in mammals. Inmice, expansion occurs in the haploid germ cells in the processof repair (35). FEN-1 effects on instability may also arise dur-ing repair.
Expression of nuclease-defective FEN-1 in human cells. Totest the hypothesis that deficient FEN-1 activity could createprecursors to expansion, we created a stable human cell linetransformed with nuclease-defective FEN-1 (54, 55) (Fig. 2A).
Normal levels of wt enzyme are present, but the mutant canbind and interfere with processing by the wt enzyme at someflaps.
SK-N-MC cells were stably transformed with an expressionvector containing the cDNA for nuclease-deficient FEN-1 (54,55) fused to an HA tag (Fig. 2B). The mutant FEN-1 containsa single aspartate-to-alanine change (D181A) in its nucleasedomain (Fig. 2A) (see Materials and Methods). This aminoacid change does not alter binding of FEN-1 to its DNA sub-strate but does eliminate the endonuclease catalytic activitythere (54, 55). Stable transformants contained wt and nucle-ase-defective FEN-1 with structure-dependent affinity for dis-placed strands. wt FEN-1 can efficiently cleave at the flapjunction, leaving a substrate for DNA ligase (Fig. 2C) (27).However, binding of the nuclease-deficient FEN-1 at the sub-strate can cause failure to cleave the flap and thereby leave aprecursor either to expansion or to a strand break (Fig. 2D).The coexpression of the mutant FEN-1 in the SK-N cells wasexpected to produce a competition with wt enzyme for bindingto the flap template (Fig. 2D).
Transformed cells were analyzed for exogenous protein ex-pression (Fig. 2B) and FEN-1 nuclease activity (Fig. 2E). Im-munoblot analysis using anti-HA antibody showed that a pro-tein of the appropriate size (the predicted mass of FEN-HA
FIG. 2. Stable transformation of SK-N-MC human neuroblastoma cells with wt or nuclease-defective FEN-1. (A) A single amino acid changefrom aspartate to alanine at amino acid 181 (D181A) in the nuclease domain of FEN-1 renders enzymes nuclease defective but does not alterbinding properties (54, 55). (B) Expression of exogenous (HA-tagged) FEN-1 in SK-N-MC cells. S, parental SK-N-MC. Numbers indicateindividual isolated colonies. Cell extracts were analyzed by immunoblotting. Proteins separated by sodium dodecyl sulfate gel electrophoresis weretransferred to nitrocellulose and probed with anti-HA antibody. (C) Binding and cleavage by wt FEN-1. The structure-specific nuclease clipsdisplaced strands at the three-way junction, leaving a substrate for ligation. (D) Mutant FEN-D181A binds normally but does not cleave, so thateven in the presence of wt enzyme some flaps may not be cleaved. Uncleaved flaps may be ligated, leading to expansion (bottom left panel), oruncleaved flaps may trigger strand break and/or subsequent repair (bottom right panel) (50). (E) FEN-1 activity in nuclear extracts fromtransformed cells. The 5� 32P-labeled flap substrate (top panel) was incubated with extract prepared from stably transformed cells or fromSK-N-MC parent cells and resolved from products on denaturing gels. Numbers indicate individual isolated cell lines. Products were 19 and 21 bp(arrows). �, BSA replaces nuclear extract. (F) Survival of SK-N-MC cells cotransformed with FEN and GFP expression plasmids. A plot of averagenumbers per field � SEM of fluorescent cells is shown. Green cells were counted in 10 fields. Tests of averages indicate a statistically significantdifference between D181A-transformed and parental SK-N-MC at each time point (P 0.002).
6066 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

fusion protein is 44,164 Da) was detected in whole-cell extractfrom hygromycin-resistant cells but not in control parent cells(SK-N) (Fig. 2B). These data confirmed that the mutant en-zyme was efficiently expressed in the human cell lines. Thelevel of wt enzyme appears to be regulated in cells, since levelsof the mutant enzyme were consistently higher than those ofthe exogenous wt enzyme in independent clones (Fig. 2B).
We tested extracts from stably transformed SK-N-MC cellsto investigate whether expression of the nuclease-deficient
FEN-1 inhibited flap processing. FEN-1 activity was measuredin vitro by incubating a Flap-1 substrate (Fig. 2E, top panel) inthe presence or absence of nuclear extract from the trans-formed cells (see Materials and Methods). FEN-1 cleaves ateither base adjacent to the junction of the displaced strand,leaving fragments of 19 and 21 nt (Fig. 2E) (24, 60). FEN-1D181A binds as well as the wt but does not cleave (54–56).
If nuclease-defective FEN-1 in extracts imparted a domi-nant-negative effect, then its presence might substantiallylower FEN-1 cleavage activity by binding to substrate andinterfering with processing by endogenous enzymes. Despiterobust expression of the mutant FEN-1 enzyme (Fig. 2B),however, we found no apparent decrease in FEN-1 cleavageactivity in the model template in any of the D181A-trans-formed cells compared to levels in the parent SK-N-MC (Fig.2E). Although contribution of another endonuclease to thetotal activity cannot be ruled out, the pattern of cleavage isidentical to that of FEN-1 previously reported in both purifiedform and in cell lysates (24, 60). Therefore, the concentrationof normal enzyme in vitro is sufficient to carry out cleavage inthe presence of nuclease-defective FEN-1. In vivo, however,FEN-1 must distribute throughout the genome and particularsites might be sensitive to the presence of the nuclease-defec-tive enzyme. Therefore, we tested whether expression of themutant FEN-1 caused a selective and/or subtle phenotype invivo.
Human cells expressing nuclease-defective FEN-1 are sus-ceptible to cell death. We found that expression of the nucle-ase-deficient FEN-1 impaired survival. To measure the effectsof nuclease-defective FEN-1 in human cells, we cotransformedSK-N-MC cells with GFP and FEN-1 expression vectors andmonitored the survival of green cells. For transformants ex-pressing the exogenous wt enzyme, we found no obvious effecton cell survival (Fig. 2F). The number of green cells in the FENwt culture was similar to that in the SK-N-MC culture as lateas 72 h after transfection. In contrast, expression of the nucle-ase-defective FEN-1 rendered cells more susceptible to deathas early as 20 h after transfection, as indicated by the lower
TABLE 2. Summary of microsatellites tested in SK-N cellsa
Locusb Repeatunit
No. ofrepeatunits
(GenBank)
No. ofinterrup-
tionsc
Lengthof longest
uninterruptedtract (repeat
units)
Repeatlength ofalleles in
SK-Nd
(repeatunits)
Alteredin D181A
cells
dyn CAG 10 1 10 10, 10 NoHD CAG 21 0 21 23, 26 YesSca1 CAG 30 2 15 NDe NoTBP CAG 38 4 18 37, 38 NoACTC CA 25 0 25 24, 26 YesD6S264 CA 20 1 17 ND NoD6S305 CA 21 0 21 14, 13 No
a Repeat regions at each of the sites indicated were amplified and sequencedin SK-N-MC parent and FEN-transformed cells.
b dyn, prodynorphin; Sca1, spinocerebellar ataxia type 1 gene; TBP, TATAbinding protein; ACTC, D6S264, and D6S305, CA microsatellite markers (20).
c Imperfections in the repeating units were sometimes present, and the num-ber of these at each locus as well as the length of the longest uninterrupted tractis indicated.
d The length as determined by amplification in SK-N-MC cells at some locidiffered from that of the GenBank sequence.
e ND, not determined.
FIG. 2—Continued.
VOL. 23, 2003 FEN-1 DEFICIENCY CAUSES REPEAT INSTABILITY 6067
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

number of GFP-positive cells in cultures expressing nuclease-defective FEN-1 (Fig. 2F). Thus, the nuclease-defective en-zyme caused the increase in cell death.
Expression of nuclease-defective FEN D181A causes repeatinstability. We hypothesize that site-specific failure to processflaps can cause repeat instability (Fig. 2D). The nuclease-de-fective FEN D181A should bind at some flaps, thereby inter-fering with cleavage by the endogenous wt enzyme and leavinga precursor for expansion (Fig. 2D). We tested whether ex-pression of the nuclease-deficient enzyme can alter the stabilityof repeat tracts of several endogenous loci (Table 2) (18, 20)(see Materials and Methods). Two of the examined loci (Sca1and HD) are associated with trinucleotide repeat diseases, andthree of the loci (ACTC, D6S305, and D6S264) have beenshown to be unstable in colon tumors (20). In normal cells,however, all of these microsatellites are stable (as is confirmedby their usefulness as highly informative markers for geneticanalysis) (34).
We analyzed DNA from parental SK-N-MC cultures, five
independent FEN wt-transformed cultures, and five indepen-dent D181A-transformed cultures. Cells were not treated withdrugs nor was any selection applied. All cells in the populationwere genetically identical at the start of the experiment. There-fore, detection of a change originating in a single cell would besignificant, since it must be distinguished from the backgroundof the population with the parental allele. A change in repeatlength would not be expected to confer selective advantage toa cell. Therefore, we relied on the sensitivity of GeneScananalysis to detect changes (35, 36) (Fig. 1C).
In the GeneScan method, repeat-containing regions arePCR amplified using one primer that is fluorescein labeled.The lengths of DNA products can be determined after reso-lution on denaturing polyacrylamide gels, and each amplifiedproduct is seen as a peak in the GeneScan trace (35, 36)(Materials and Methods). At the HD allele, for example, sep-aration of the two prominent peaks in SK-N-MC cells indicatesthat the long and short HD alleles differ by 9 bp or threerepeats (Fig. 3A, HD [right panel]). Alterations in repeatlength cause changes in the sizes of PCR product. Since repeatlengths change by a discrete number of nucleotides, peaks thatshift position are often coincident with another peak in thetrace. Thus, repeat length changes are identified either as theappearance of a new peak or by changes in peak areas (Fig.3A; compare results for parent SK-N-MC and D181A).
Amplification of repeat-containing regions typically yields aprominent band with adjacent smaller bands (Fig. 3A) (20, 69).SK-N-MC and FEN wt-transformed cultures are indistinguish-able by microsatellite analysis. Reproducible deviations from
FIG. 3. Microsatellite instability is caused by expression of nucle-ase-deficient FEN-1. (A) GeneScan analysis of ACTC and HD alleles.Left panels, ACTC alleles amplified with primer set 1; middle panels,ACTC alleles with primer set 2; right panels, HD alleles with primerset 1; top scans, SK-N-MC cells; middle scans, FEN wt-transformedcells; bottom scans, D181A-transformed samples. Dashed lines indi-cate boundaries between two alleles, arrows indicate prominent peaks,and dots represent the secondary peak positions. Numbers indicate thelength of the amplified DNA. (B to D) Fraction of area under prom-inent peak for the indicated allele. Values were from a GeneScananalysis table. Averages � SEM are shown. A t test was used todetermine whether averages were the same (GraphPad Prism).(B) For longer HD alleles, the fraction of the allele under the prom-inent peak was determined using primer set 1 (filled symbols) orprimer set 2 (unfilled symbols). (C) For ACTC alleles, the fraction ofthe allele under the prominent peak was determined using primer set1 (filled symbols) or primer set 2 (unfilled symbols). (D) For shorterHD alleles, the fraction of the allele under the prominent peak wasdetermined using primer set 1 (filled symbols) or primer set 2 (unfilledsymbols).
6068 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.
the parental pattern were detected in D181A cultures at twotest loci, ACTC (a microsatellite within the gene encodingcardiac muscle actin) and HD (Table 2; Fig. 3A). Changingpeak areas within the GeneScan traces indicated that in theD181A sample, there were numbers of repeats different fromthose in the SK-N-MC parent or FEN wt at both loci (Fig. 3A).To confirm that the altered patterns were due to repeat-lengthchanges within the target gene, both the ACTC and HD alleleswere amplified with two different primer sets (primer 1 andprimer 2) (Fig. 3A). For both ACTC and HD, amplificationwith alternative primer sets yielded the same pattern of prod-ucts but shifted by the appropriate distance (Fig. 3A; resultsare shown for ACTC only [compare primer 1 and primer 2results]). Thus, the observed microsatellite instability withinthe PCR products was gene specific.
At both ACTC alleles and the long HD allele in D181A-transformed cultures, the ratio of the prominent peak area(Fig. 3A) is altered relative to that of the secondary peak in thetrace (Fig. 3A). Using both sets of primers (Fig. 3B to D), wecalculated the fraction under the prominent peak (most-abun-dant repeat length) for the ACTC alleles and the long HDallele in independent amplification reactions (Fig. 3B to D).The analysis demonstrates that the repeat length of one HDallele was indistinguishable among the SK-N-MC and FENwt-transformed cultures but differed significantly in FEND181A-transformed cultures (P 0.0001) (Fig. 3B to D). Both
sets of primers yielded similar results, indicating that the re-peat lengths reflected a property of the specific alleles (Fig. 3Band C). As an internal control, we found that the shorter HDallele was the same in all of the samples (Fig. 3D).
Therefore, expression of nuclease-defective FEN-1 is suffi-cient to cause microsatellite instability. An uncleaved flap maybe a direct precursor to instability and may disrupt repairactivities in transformed cells.
Expression of nuclease-deficient FEN-1 causes a reducedcapacity to correct strand breaks. Experiments with nuclease-defective FEN-1 confirmed that repair of displaced flaps isessential for genome stability (Fig. 3). Instability must, there-fore, arise from faulty repair of flaps. To test relevant process-ing events, we treated cells with DNA-damaging agents thatmight directly or indirectly create flaps. MMS increases thenumber of alkylated bases, which are typically repaired byBER (10). BER causes single-strand DNA breaks in the pro-cess of removal of alkylated bases, and this process can beFEN-1 dependent (13, 32, 33, 47, 49, 56, 64). However, in cellcultures, a high degree of MMS-mediated single-strand breakscan also result in double-strand breaks (66).
We also used cisplatin to induce strand breaks. Cisplatincreates bulky lesions that can be removed by NER (3, 10, 11,28). Cisplatin can also form interstrand cross-links that arecorrected by double-strand break repair (4, 73). After eithertreatment, survival of drug-treated cells was measured by col-ony survival assay (Materials and Methods).
Expression of the exogenous wt FEN-1 and D181A FEN-1caused cells to be hypersensitive to MMS (Fig. 4A) comparedto the parent SK-N-MC cells. However, cells expressing thenuclease-defective FEN-1 did not differ in MMS sensitivityrelative to the cells exogenously expressing wt FEN-1 (Fig.4A). The activity of FEN-1 is regulated by its direct interactionwith many cellular enzymes, including proliferating cell nuclearantigen, a DNA polymerase , and ε processivity factor (14, 19,22, 31, 38, 39, 56, 63, 64, 68); the transcriptional coregulatorand acetylase P300 (25); and the WRN helicase (6). Further-more, the cell cycle regulator p21waf1/CIP shares a proliferatingcell nuclear antigen interaction domain with FEN-1 (64, 67),and the competition between FEN-1 and p21 modulates theresponse to DNA damage (64). That cells expressing wt andnuclease-defective FEN-1 were equally susceptible to MMSsuggests that the levels of exogenous protein (expressed undercontrol of the cytomegalovirus promoter) can disrupt the pro-tein-protein interactions required for proper cellular metabo-lism.
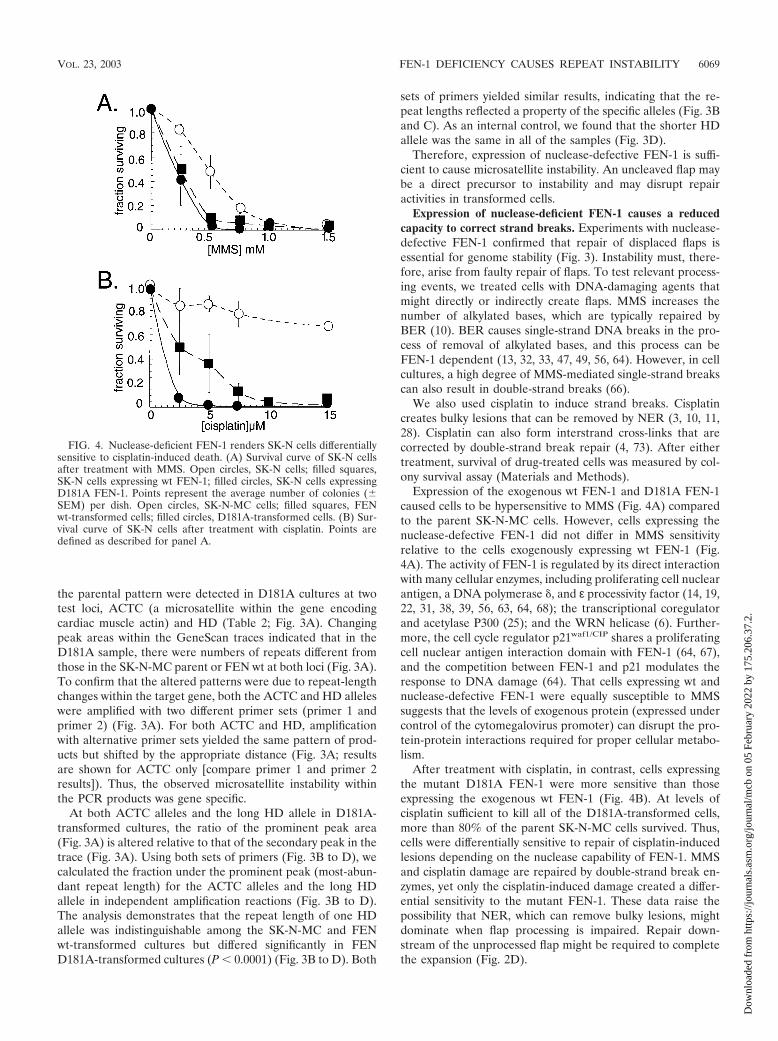
After treatment with cisplatin, in contrast, cells expressingthe mutant D181A FEN-1 were more sensitive than thoseexpressing the exogenous wt FEN-1 (Fig. 4B). At levels ofcisplatin sufficient to kill all of the D181A-transformed cells,more than 80% of the parent SK-N-MC cells survived. Thus,cells were differentially sensitive to repair of cisplatin-inducedlesions depending on the nuclease capability of FEN-1. MMSand cisplatin damage are repaired by double-strand break en-zymes, yet only the cisplatin-induced damage created a differ-ential sensitivity to the mutant FEN-1. These data raise thepossibility that NER, which can remove bulky lesions, mightdominate when flap processing is impaired. Repair down-stream of the unprocessed flap might be required to completethe expansion (Fig. 2D).
FIG. 4. Nuclease-deficient FEN-1 renders SK-N cells differentiallysensitive to cisplatin-induced death. (A) Survival curve of SK-N cellsafter treatment with MMS. Open circles, SK-N cells; filled squares,SK-N cells expressing wt FEN-1; filled circles, SK-N cells expressingD181A FEN-1. Points represent the average number of colonies (�SEM) per dish. Open circles, SK-N-MC cells; filled squares, FENwt-transformed cells; filled circles, D181A-transformed cells. (B) Sur-vival curve of SK-N cells after treatment with cisplatin. Points aredefined as described for panel A.
VOL. 23, 2003 FEN-1 DEFICIENCY CAUSES REPEAT INSTABILITY 6069
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.
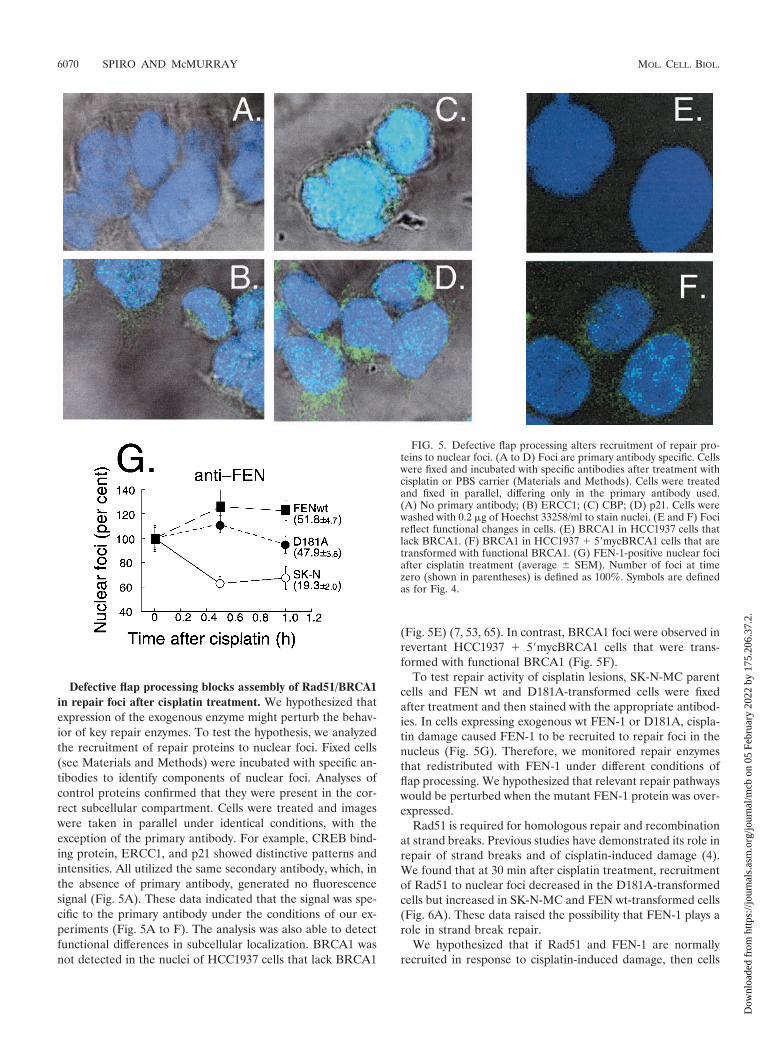
Defective flap processing blocks assembly of Rad51/BRCA1in repair foci after cisplatin treatment. We hypothesized thatexpression of the exogenous enzyme might perturb the behav-ior of key repair enzymes. To test the hypothesis, we analyzedthe recruitment of repair proteins to nuclear foci. Fixed cells(see Materials and Methods) were incubated with specific an-tibodies to identify components of nuclear foci. Analyses ofcontrol proteins confirmed that they were present in the cor-rect subcellular compartment. Cells were treated and imageswere taken in parallel under identical conditions, with theexception of the primary antibody. For example, CREB bind-ing protein, ERCC1, and p21 showed distinctive patterns andintensities. All utilized the same secondary antibody, which, inthe absence of primary antibody, generated no fluorescencesignal (Fig. 5A). These data indicated that the signal was spe-cific to the primary antibody under the conditions of our ex-periments (Fig. 5A to F). The analysis was also able to detectfunctional differences in subcellular localization. BRCA1 wasnot detected in the nuclei of HCC1937 cells that lack BRCA1
(Fig. 5E) (7, 53, 65). In contrast, BRCA1 foci were observed inrevertant HCC1937 � 5�mycBRCA1 cells that were trans-formed with functional BRCA1 (Fig. 5F).
To test repair activity of cisplatin lesions, SK-N-MC parentcells and FEN wt and D181A-transformed cells were fixedafter treatment and then stained with the appropriate antibod-ies. In cells expressing exogenous wt FEN-1 or D181A, cispla-tin damage caused FEN-1 to be recruited to repair foci in thenucleus (Fig. 5G). Therefore, we monitored repair enzymesthat redistributed with FEN-1 under different conditions offlap processing. We hypothesized that relevant repair pathwayswould be perturbed when the mutant FEN-1 protein was over-expressed.
Rad51 is required for homologous repair and recombinationat strand breaks. Previous studies have demonstrated its role inrepair of strand breaks and of cisplatin-induced damage (4).We found that at 30 min after cisplatin treatment, recruitmentof Rad51 to nuclear foci decreased in the D181A-transformedcells but increased in SK-N-MC and FEN wt-transformed cells(Fig. 6A). These data raised the possibility that FEN-1 plays arole in strand break repair.
We hypothesized that if Rad51 and FEN-1 are normallyrecruited in response to cisplatin-induced damage, then cells
FIG. 5. Defective flap processing alters recruitment of repair pro-teins to nuclear foci. (A to D) Foci are primary antibody specific. Cellswere fixed and incubated with specific antibodies after treatment withcisplatin or PBS carrier (Materials and Methods). Cells were treatedand fixed in parallel, differing only in the primary antibody used.(A) No primary antibody; (B) ERCC1; (C) CBP; (D) p21. Cells werewashed with 0.2 �g of Hoechst 33258/ml to stain nuclei. (E and F) Focireflect functional changes in cells. (E) BRCA1 in HCC1937 cells thatlack BRCA1. (F) BRCA1 in HCC1937 � 5�mycBRCA1 cells that aretransformed with functional BRCA1. (G) FEN-1-positive nuclear fociafter cisplatin treatment (average � SEM). Number of foci at timezero (shown in parentheses) is defined as 100%. Symbols are definedas for Fig. 4.
6070 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

defective in Rad51 pathway should show abnormal recruitingof FEN-1. We, therefore, tested whether FEN-1 was recruitedto repair foci in the HCC1937 cell line, which lacks a functionalBRCA1 (7, 53, 65). In BRCA1-deficient cells treated withcisplatin, Rad51-containing complexes do not assemble andDNA repair is severely impaired (4). In HCC1937 cells, thenumber of FEN-1-containing foci decreased after cisplatintreatment (Fig. 6B). However, recruitment of FEN-1 to nu-clear foci was restored after cisplatin treatment of the revertantHCC1937 � 5�mycBRCA1 line, which expresses a functionalBRCA1 transgene (Fig. 6B). Thus, the presence of wt FEN-1in repair foci was restored when Rad51 assembly was compe-tent. These data suggested that FEN-1 normally plays a role inBRCA1/RAD51-dependent repair but that impaired flap pro-cessing invoked an alternative pathway to compensate for aRad51 deficit.
A substantial body of evidence has shown that NER canremove cisplatin intrastrand cross-links (3, 10, 11, 28). There-fore, we tested whether key components of the NER pathwaywere recruited to repair foci when flap processing was im-paired. ERCC1 forms a nuclease with XPF that cleaves 5� to abulky lesion (3, 10, 11). We found that within 30 min of cis-platin treatment, there was a marked increase in the number ofERCC1 nuclear foci in D181A-transformed cells (Fig. 6D and5F). Under the same conditions, cells overexpressing the wt
FEN-1 displayed little to no change (Fig. 6C and 5F). It ap-peared that some components of the NER pathway were re-cruited to repair foci when flap processing was impaired. Forcells deficient in NER (XPC671 cells) (8) (Fig. 6D), in con-trast, there was no effect on FEN-1 focus formation relative tocontrol cells (Fig. 6D). The staining of ERCC1 appeared to bespecific. We tested other components of the NER pathway.For XPG, focus formation in D181A-transformed cells differedfrom that of FEN wt-transformed or from parental SK-N-MCcells but with a somewhat different pattern from that seen withERCC1 (data not shown). The percentage of XPG foci inD181A-transformed cells decreased (56.2% � 2.0%) relativeto parent SK-N cells (74.9% � 3.5%) and to FEN wt-trans-formed cells (99.9% � 5.4%) within 30 min of cisplatin treat-ment.
DISCUSSION
We have shown that in human cells, expression of nuclease-defective FEN-1 has profound effects, including instability atendogenous repetitive sites and alterations in the response ofRAD 51. Inefficient flap processing appears to prevent Rad51-mediated repair and may invoke components of NER to over-come the Rad51 deficit. Taken together, the data indicate thatinefficient flap processing by FEN-1 contributes to CAG insta-
FIG. 6. Defective flap processing blocks recruitment of Rad51, and loss of BRCA1 blocks recruitment of FEN-1. (A) Rad51-positive nuclearfoci (conditions and labels are defined as for Fig. 5G) after cisplatin treatment. (B) FEN-1-positive foci in HCC1937 cells that lack BRCA1 (opencircles) and in revertant HCC1937 � 5�mycBRCA1 cells (filled squares). (C) ERCC1-positive nuclear foci after cisplatin treatment. (D) FEN-1-positive foci in Rovid cells or in NER-defective XPC 671 cells (8).
VOL. 23, 2003 FEN-1 DEFICIENCY CAUSES REPEAT INSTABILITY 6071
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

bility and confirms that wt FEN-1 has a ubiquitous role inmaintaining genome stability in eukaryotes from yeast to hu-mans.
Model for nuclease-defective FEN-1-mediated instability inmammals. The data suggest a model by which FEN-1 main-tains genome stability and aberrant FEN-1 processing under-lies instability (Fig. 7). Normal processing by FEN-1 removesflaps that might give rise to instability (Fig. 3 and 7A). Al-though the mechanism of interaction is unknown, FEN-1 re-pair appears to depend on a complex comprising Rad51 andBRCA1 (Fig. 6B and 7A). Previous studies have not identifieda role for FEN-1 in double-strand break repair (56), but Rad51and BRCA1 are required for repair of cisplatin-induced DNAdamage (4, 5). We show here that repair of cisplatin damagealso requires functional FEN-1 (Fig. 4B) and that recruitmentof FEN-1 depends on BRCA1 (Fig. 6B). Furthermore, defec-tive FEN-1 nuclease activity inhibits recruitment of Rad 51 torepair foci after cisplatin treatment (Fig. 6A). Cells can survivein the presence of the nuclease-defective enzyme, but ourstudies show that its expression causes a failure to assembleRad51-containing repair complexes even in the presence of wtFEN-1. This implies that repair in the absence of intact Rad 51complexes occurs via other pathways (Fig. 5F and 6A).
The absence of flap endonuclease (Rad27) in yeast causes amutator phenotype with genome-wide instability. In trinucle-otide expansion disease patients, however, FEN-1 is not de-fective, since only repeats at the disease locus are unstable.Thus, single-site mutation in trinucleotide diseases does notreflect a general fault of the protein machinery but reflects theinability of this machinery to function normally at the repeat
site (20). Flaps comprising CAG repeats form stable hairpinswith mismatched bases in their stems that are recognized by anMsh2 complex (17, 35, 48). We and others have previouslyshown that stable hydrogen bonds within the hairpin can pre-vent FEN-1-mediated flap processing (26, 60). Therefore, atthe disease locus, FEN-1 is essentially nuclease defective andleaves a precursor for expansion (Fig. 7B). We speculate thatin disease-related expansion, the proteins that repair the pre-cursor flap are similar to those recruited during expression ofthe nuclease-defective FEN-1. If this were correct, thenRad51/BRCA1 complexes would fail to assemble at theblocked FEN-1 site (Fig. 7B) and an alternative pathway in-volving ERCC1 would be recruited under conditions in whichflap cleavage is inefficient.
The alternative repair pathway is unlikely to be canonicalNER, since XPG focus formation in D181A cells does notfollow the same pattern as that of ERCC1 within a similar timeframe after cisplatin treatment. ERCC1 has a role in NER butalso has other functions in the cell, notably in strand breakrepair (1). In fact, evidence for a non-NER pathway that in-volves ERCC1 has been recently reported (12). Chinese ham-ster ovary lines defective in individual components of NERwere unable to repair both cisplatin-induced interstrand andintrastrand cross-links. ERCC1- and XPF-deficient lines wereextremely sensitive to cisplatin damage, however, in contrast tolines deficient in other components of NER. Under these con-ditions, cisplatin adducts did not appear to involve the forma-tion of DNA double-strand breaks. EFCC1-deficient cells can-not support NER. However, the high level of cisplatinsensitivity of ERCC1- and XPF-deficient cells indicates a de-fect in another pathway, possibly single-strand annealing.
The alternative pathway that gives rise to trinucleotide in-stability is unclear. However, one of two mechanisms mayunderlie the mutation. A repair complex may be recruited butfail to excise the hairpin, in which case the hairpin is incorpo-rated into the DNA (Fig. 7B). Alternatively, a repair complexmay succeed in excising the hairpin; then extra repeats areincorporated after slippage during repair synthesis (Fig. 7B).Further studies employing multiple repair enzymes will berequired to firmly establish a mechanism.
ACKNOWLEDGMENTS
We thank R. Kucherlapati (Harvard Medical School) for FEN-1heterozygous knockout mice, J. Chen (Mayo Clinic) for HCC1937 celllines, M. S. Park (Los Alamos National Laboratory) for FEN-1cDNAs, and V. S. Pankratz (Mayo Clinic) for critical comments ondata analysis.
This work was supported by the Mayo Foundation, the HereditaryDisease Foundation, the National Institutes of Health (DK 43694 andMH56207), and the National Science Foundation (IBN 9728120 [toC.T.M.]).
REFERENCES
1. Adair, G. M., R. L. Rolig, D. Moore-Faver, M. Dabelshansky, J. H. Wilson,and R. S. Nairn. 2000. Role of ERCC1 in removal of long non-homologoustails during targeted non-homologous recombination. EMBO J. 19:5552–5561.
2. Andrews, N. C., and D. V. Faller. 1991. A rapid micropreparation techniquefor extraction of DNA-binding proteins from limiting numbers of mamma-lian cells. Nucleic Acids Res. 19:2499.
3. Araujo, S. J., F. Tirode, F. Coin, H. Pospiech, J. E. Syvaoja, M. Stucki, U.Hubscher, J. M. Egly, and R. D. Wood. 2000. Nucleotide excision repair ofDNA with recombinant human proteins: definition of the minimal set offactors, active forms of TFIIH, and modulation by CAK. Genes Dev. 14:349–359.
FIG. 7. A possible model to explain FEN-1-dependent genome sta-bility and how defective flap processing causes triplet expansion.(A) Displaced strand is cleaved to leave precursor to ligation. wtFEN-1 is required for formation of Rad 51/BRCA1 complexes duringnormal flap processing. The question mark indicates that the mode ofinteraction of these components with FEN-1 and the sequence of thebinding events are unknown. (B) A stable hairpin forms at the CAGrepeat with mismatched base pairs in the stem. The stable hairpinblocks FEN-1 processing (Cleavage block) and formation of Rad51/BRCA1 repair complex and recruits a mismatch repair complex. De-fective flap processing causes recruitment of another repair pathway toexcise the hairpin. Expansion occurs in the process.
6072 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

4. Bhattacharyya, A., U. S. Ear, B. H. Koller, R. R. Weichselbaum, and D. K.Bishop. 2000. The breast cancer susceptibility gene BRCA1 is required forsubnuclear assembly of Rad51 and survival following treatment with theDNA cross-linking agent cisplatin. J. Biol. Chem. 275:23899–23903.
5. Bishop, D. K., U. Ear, A. Bhattacharyya, C. Calderone, M. Beckett, R. R.Weichselbaum, and A. Shinohara. 1998. XRCC3 is required for assembly ofRad51 complexes in vivo. J. Biol. Chem. 273:21482–21488.
6. Brosh, R. M., C. von Kobbe, J. A. Sommers, P. Karmakar, P. L. Opresko, J.Piotrowski, I. Dianova, G. L. Dianov, and V. A. Bohr. 2001. Werner syn-drome protein interacts with human flap endonuclease 1 and stimulates itscleavage activity. EMBO J. 20:5791–5801.
7. Chen, J., D. P. Silver, D. Walpita, S. B. Cantor, A. F. Gazdar, G. Tomlinson,F. J. Couch, B. L. Weber, T. Ashley, D. M. Livingston, and R. Scully. 1998.Stable interaction between the products of the BRCA1 and BRCA2 tumorsuppressor genes in mitotic and meiotic cells. Mol. Cell 2:317–328.
8. Cleaver, J. E., L. H. Thompson, A. S. Richardson, and J. C. States. 1999. Asummary of mutations in the UV-sensitive disorders: xeroderma pigmento-sum, Cockayne syndrome, and trichothiodystrophy. Hum. Mutat. 14:9–22.
9. Daly, L. E. and G. J. Bourke. 2000. Interpretation and uses of medicalstatistics, 5th ed. Blackwell Science Ltd., Oxford, United Kingdom.
10. de Boer, J., and J. H. J. Hoeijmakers. 2000. Nucleotide excision repair andhuman syndromes. Carcinogenesis 21:453–460.
11. de Laat, W. L., N. G. J. Jaspers, and J. H. J. Hoeijmakers. 1999. Molecularmechanism of nucleotide excision repair. Genes Dev. 13:768–785.
12. DeSilva, I. U., P. J. McHugh, P. H. Clingen, and J. A. Hartley. 2002. Defectsin intrastrand crosslinks uncoupling do not account for extreme sensitivity ofERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 30:3848–3856.
13. Dianova, I. I., V. A. Bohr, and G. L. Dianov. 2001. Interaction of human APendonuclease 1 with flap endonuclease 1 and proliferating cell nuclear an-tigen involved in long-patch base excision. Biochemistry 40:12639–12644.
14. Frank, G., J. Qiu, L. Zheng, and B. Shen. 2001. Stimulation of eukaryoticflap endonuclease-1 activities by proliferating cell nuclear antigen (PCNA) isindependent of its in vitro interaction via a consensus PCNA binding region.J. Biol. Chem. 276:36295–36302.
15. Freudenreich, C. H., S. M. Kantrow, and V. A. Zakian. 1998. Expansion andlength-dependent fragility of CTG repeats in yeast. Science 279:853–856.
16. Freudenreich, C. H., J. B. Stavenhagen, and V. A. Zakain. 1997. Stability ofa CTG/CAG trinucleotide repeat in yeast is dependent on its orientation inthe genome. Mol. Cell. Biol. 17:2090–2098.
17. Gacy, A. M., G. Goellner, N. Juranic, S. Macura, and C. T. McMurray. 1995.Trinucleotide repeats that expand in human disease form hairpin structuresin vitro. Cell 81:533–540.
18. Gacy, A. M., G. M. Goellner, C. Spiro, X. Chen, G. Gupta, E. M. Bradbury,R. B. Dyer, M. J. Mikesell, J. Z. Yao, A. J. Johnson, A. Richter, S. B.Melancon, and C. T. McMurray. 1998. GAA instability in Friedreich’s ataxiashares a common, DNA-directed and intraallelic mechanism with othertrinucleotide diseases. Mol. Cell 1:583–593.
19. Gary, R., M. S. Park, J. P. Nolan, H. L. Cornelius, O. G. Kozyreva, H. T.Tran, K. S. Lobachev, M. A. Resnick, and D. A. Gordenin. 1999. A novel rolein DNA metabolism for the binding of FEN1/Rad27 to PCNA and implica-tions for genetic risk. Mol. Cell. Biol. 19:5373–5382.
20. Goellner, G. M., D. Tester, S. Thibodeau, E. Almqvist, Y. P. Goldberg, M. R.Hayden, and C. T. McMurray. 1997. Different mechanisms underlie DNAinstability in Huntington disease and colorectal cancer. Am. J. Hum. Genet.60:879–890.
21. Goldberg, Y. P., C. T. McMurray, J. Zeisler, E. Almqvist, D. Sillence, F.Richards, A. M. Gacy, J. Buchanan, H. Telenius, and M. R. Hayden. 1995.Increased instability of intermediate alleles in families with sporadic Hun-tington disease compared to similar sized intermediate alleles in the generalpopulation. Hum. Mol. Genet. 4:1911–1918.
22. Greene, A. L., J. R. Snipe, D. A. Gordenin, and M. A. Resnick. 1999.Functional analysis of human FEN1 in Saccharomyces cerevisiae and its rolein genome stability. Hum. Mol. Genet. 8:2263–2273.
23. Hansen, R. J., E. C. Friedberg, and M. S. Reagan. 2000. Sensitivity of a S.cerevisiae RAD27 deletion mutant to DNA-damaging agents and in vivocomplementation by the human FEN-1 gene. Mutat. Res. 461:243–248.
24. Harrington, J. J., and M. R. Lieber. 1994. The characterization of a mam-malian structure-specific DNA endonuclease. EMBO J. 13:1235–1246.
25. Hasan, S., M. Stucki, P. O. Hassa, R. Imhof, P. Gehrig, P. Hunziker, U.Hubscher, and M. O. Hottiger. 2001. Regulation of human flap endonucle-ase-1 activity by acetylation through the transcriptional coactivator p300.Mol. Cell 7:1221–1231.
26. Henricksen, L. A., S. Tom, Y. Liu, and R. A. Bambara. 2000. Inhibition offlap endonuclease 1 by flap secondary structure and relevance to repeatsequence expansion. J. Biol. Chem. 275:16420–16427.
27. Henricksen, L. A., J. Veeraraghavan, D. R. Chafin, and R. A. Bambara. 2002.DNA ligase I competes with FEN1 to expand repetitive DNA sequences invitro. J. Biol. Chem. 277:22361–22369.
28. Jamieson, E. R., and S. J. Lippard. 1999. Structure, recognition, and pro-cessing of cisplatin-DNA adducts. Chem. Rev. 99:2467–2498.
29. Jin, Y. H., R. Obert, P. M. J. Burgers, T. A. Kunkel, M. A. Resnick, and D. A.Gordenin. 2001. The 3�35� exonuclease of DNA polymerase delta can
substitute for the 5� flap endonuclease Rad27/Fen1 in processing Okazakifragments and preventing genome instability. Proc. Natl. Acad. Sci. USA98:5122–5127.
30. Johnson, R. E., G. K. Kovvali, L. Prakash, and S. Prakash. 1995. Require-ment of the yeast RTH1 5� to 3� exonuclease for the stability of simplerepetitive DNA. Science 269:238–240.
31. Kim, I.-S., M.-Y. Lee, I.-H. Lee, S.-L. Shin, and S.-Y. Lee. 2000. Geneexpression of flap endonuclease-1 during cell proliferation and differentia-tion. Biochim. Biophys. Acta 1496:333–340.
32. Kim, K., S. Biade, and Y. Matsumoto. 1998. Involvement of flap endonu-clease 1 in base excision DNA repair. J. Biol. Chem. 273:8842–8848.
33. Klungland, A., and T. Lindahl. 1997. Second pathway for completion ofhuman DNA base-excision repair: reconstitution with purified proteins andrequirement for DNase IV (FEN1). EMBO J. 16:3341–3348.
34. Korf, B. R., and R. A. Pagon. 2001. Overview of molecular genetic diagnosis,p. 9.2.1–9.2.6. In N. C. Dracopoli, J. L. Haines, B. R. Korf, D. T. Moir, C. C.Morton, C. E. Seidman, J. G. Seidman, and D. R. Smith (ed.), Currentprotocols in human genetics. John Wiley & Sons, New York, N.Y.
35. Kovtun, I. V., and C. T. McMurray. 2001. Trinucleotide expansion in haploidgerm cells by gap repair. Nat. Genet. 27:407–411.
36. Kovtun, I. V., T. M. Therneau, and C. T. McMurray. 2000. Gender of theembryo contributes to CAG instability in transgenic mice containing a Hun-tington’s disease gene. Hum. Mol. Genet. 9:2767–2775.
37. Kucherlapati, M., K. Yang, M. Kuraguchi, J. Zhao, M. Lia, J. Heyer, M. F.Kane, K. Fan, R. Russell, A. M. C. Brown, B. Kneitz, W. Edelmann, R. D.Kolodner, M. Lipkin, and R. Kucherlapati. 2002. Haploinsufficiency of Flapendonuclease (Fen1) leads to rapid tumor progression. Proc. Natl. Acad. Sci.USA 99:9924–9929.
38. Li, X., J. Li, J. Harrington, M. R. Lieber, and P. M. J. Burgers. 1995. Laggingstrand DNA synthesis at the eukaryotic replication fork involves binding andstimulation of FEN-1 by proliferating cell nuclear antigen. J. Biol. Chem.270:22109–22112.
39. Lieber, M. R. 1997. The FEN-1 family of structure-specific nucleases ineukaryotic DNA replication, recombination and repair. Bioessays 19:233–240.
40. Mangiarini, L., K. Sathasivam, M. Seller, B. Cozens, A. Harper, C. Heth-erington, M. Lawton, Y. Trottier, H. Lehrach, S. W. Davies, and G. P. Bates.1996. Exon 1 of the HD gene with an expanded CAG repeat is sufficient tocause progressive neurological phenotype in transgenic mice. Cell 87:493–506.
41. Manley, K., T. L. Shirley, L. Flaherty, and A. Messer. 1999. Msh2 deficiencyprevents in vivo somatic instability of the CAG repeat in Huntington diseasetransgenic mice Nat. Genet. 23:471–473.
42. Maurer, D. J., B. L. O’Callaghan, and D. M. Livingston. 1996. Orientationdependence of trinucleotide CAG repeat instability in Saccharomyces cer-evisiae. Mol. Cell. Biol. 16:6617–6622.
43. McMurray, C. T. 1999. DNA secondary structure: a common and causativefactor for expansion in human disease. Proc. Natl. Acad. Sci. USA 96:1823–1825.
44. Miret, J. J., L. Pessoa-Brandao, and R. S. Lahue. 1997. Instability of CAGand CTG trinucleotide repeats in Saccharomyces cerevisiae. Mol. Cell. Biol.17:3382–3387.
45. Moreau, S., E. A. Morgan, and L. S. Symington. 2001. Overlapping functionsof the Saccharomyces cerevisiae Mre11, Exo1 and Rad27 nucleases in DNAmetabolism. Genetics 159:1423–1433.
46. Negritto, M. C., J. Qiu, D. O. Ratay, B. Shen, and A. M. Bailis. 2001. Novelfunction of Rad27 (FEN-1) in restricting short-sequence recombination.Mol. Cell. Biol. 21:2349–2358.
47. Pascucci, B., G. Maga, U. Hubscher, M. Bjoras, E. Seeberg, I. D. Hickson, G.Villani, C. Giordano, L. Cellai, and E. Dogliotti. 2002. Reconstitution of thebase excision repair pathway for 7,8-dihydro-8-oxoguanine with purified hu-man proteins. Nucleic Acids Res. 30:2124–2130.
48. Pearson, C. E., A. Ewel, S. Acharya, R. A. Fishel, and R. R. Sinden. 1997.Human Msh2 binds to trinucleotide repeat DNA structures associated withneurodegenerative diseases. Hum. Mol. Genet. 6:1117–1123.
49. Qiu, J., X. Li, G. Frank, and B. Shen. 2001. Cell cycle-dependent and DNAdamage-inducible nuclear localization of FEN-1 nuclease is consistent withits dual functions in DNA replication and repair. J. Biol. Chem. 276:4901–4908.
50. Richard, G.-F., G. M. Goellner, C. T. McMurray, and J. E. Haber. 2000.Recombination-induced CAG trinucleotide repeat expansions in yeast in-volve the MRE11/RAD50/XRS2 complex. EMBO J. 19:2381–2390.
51. Sarkar, P. S., H. C. Chang, F. B. Boudi, and S. Reddy. 1998. CTG repeatsshow bimodal amplification in E. coli. Cell 95:531–540.
52. Schweitzer, J. K., and D. M. Livingston. 1998. Expansions of CAG repeattracts are frequent in a yeast mutant defective in Okazaki fragment matu-ration. Hum. Mol. Genet. 7:69–74.
53. Scully, R., S. Ganesan, K. Vlasakova, J. Chen, M. Socolovsky, and D. M.Livingston. 1999. Genetic analysis of BRCA1 function in a defined tumorcell line. Mol. Cell 4:1093–1099.
54. Shen, B., J. P. Nolan, L. A. Sklar, and M. S. Park. 1996. Essential amino
VOL. 23, 2003 FEN-1 DEFICIENCY CAUSES REPEAT INSTABILITY 6073
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.

acids for substrate binding and catalysis of human flap endonuclease 1.J. Biol. Chem. 271:9173–9176.
55. Shen, B., J. P. Nolan, L. A. Sklar, and M. S. Park. 1997. Functional analysisof point mutations in human flap endonuclease-1 active site. Nucleic AcidsRes. 25:3332–3338.
56. Shibata, Y., and T. Nakamura. 2002. Defective flap endonuclease 1 activityin mammalian cells is associated with impaired DNA repair and prolongedS phase delay. J. Biol. Chem. 277:746–754.
57. Shivji, M. K. K., V. N. Podust, U. Hubscher, and R. D. Wood. 1995. Nucle-otide excision repair DNA synthesis by DNA polymerase ε in the presenceof PCNA, RFC and RPA. Biochemistry 34:5011–5017.
58. Spiro, C., D. P. Bazett-Jones, X. Wu, and C. T. McMurray. 1995. DNAstructure determines protein binding and transcriptional efficiency of theproenkephalin cAMP-responsive enhancer. J. Biol. Chem. 270:27702–27710.
59. Spiro, C., and C. T. McMurray. 1999. Footprint analysis of DNA-proteincomplexes in vitro and in vivo, p. 27–62. In D. S. Latchman (ed.), Transcrip-tion factors: a practical approach, 2nd edition. Oxford University Press,Oxford, United Kingdom.
60. Spiro, C., R. Pelletier, M. L. Rolfsmeier, M. J. Dixon, R. S. Lahue, G. Gupta,M. S. Park, X. Chen, S. V. S. Mariappan, and C. T. McMurray. 1999.Inhibition of FEN-1 processing by DNA secondary structure at trinucleotiderepeats. Mol. Cell 4:1079–1085.
61. Symington, L. S. 1998. Homologous recombination is required for the via-bility of rad27 mutants. Nucleic Acids Res. 26:5589–5595.
62. Tishkoff, D. X., N. Filosi, G. M. Gaida, and R. D. Kolodner. 1997. A novelmutation avoidance mechanism dependent on S. cerevisiae RAD27 is distinctfrom DNA mismatch repair. Cell 88:253–263.
63. Tom, S., L. A. Henricksen, and R. A. Bambara. 2000. Mechanism wherebyproliferating cell nuclear antigen stimulates flap endonuclease 1. J. Biol.Chem. 275:10498–10505.
64. Tom, S., T. A. Ranalli, V. N. Podust, and R. A. Bambara. 2001. Regulatoryroles of p21 and apurinic/apyrimidinic endonuclease 1 in base excision re-pair. J. Biol. Chem. 276:48781–48789.
65. Tomlinson, G. E., T. T. Chen, V. A. Stastny, A. K. Virmani, M. A. Spillman,V. Tonk, J. L. Blum, N. R. Schneider, I. I. Wistuba, J. W. Shay, J. D. Minna,and A. F. Gazdar. 1998. Characterization of a breast cancer cell line derivedfrom a germ-line BRCA1 mutation carrier. Cancer Res. 58:3237–3242.
66. Tsubouchi, H., and H. Ogawa. 2000. Exo1 roles for repair of DNA double-strand breaks and meiotic crossing over in Saccharomyces cerevisiae. Mol.Biol. Cell 11:2221–2233.
67. Warbrick, E., D. P. Lane, D. M. Glover, and L. S. Cox. 1997. Homologousregions of Fen1 and p21Cip1 compete for binding to the same site on PCNA:a potential mechanism to co-ordinate DNA replication and repair. Onco-gene 14:2313–2321.
68. Warbrick, E., P. J. Coates, and P. A. Hall. 1998. FEN1 expression: a novelmarker for cell proliferation. J. Pathol. 186:319–324.
69. Wellington, C. L., L. M. Ellerby, C. A. Gutekunst, D. Rogers, S. Warby, R. K.Graham, O. Loubser, J. van Raamsdonk, R. Singaraja, Y. Yang, J. Gafni, D.Bredesen, S. M. Hersch, B. R. Leavitt, S. Roy, D. W. Nicholson, and M. R. J.Hayden. 2002. Caspase cleavage of mutant huntingtin precedes neurodegen-eration in Huntington’s disease. J. Neurosci. 22:7862–7872.
70. Wu, X., T. E. Wilson, and M. R. Lieber. 1999. A role for FEN-1 in nonho-mologous end joining: the order of strand annealing and nucleolytic pro-cessing events. Proc. Natl. Acad. Sci. USA 96:1303–1308.
71. Xie, Y., Y. Liu, J. L. Argueso, L. A. Henricksen, H.-I. Kao, R. A. Bambara,and E. Alani. 2001. Identification of rad27 mutations that confer differentialdefects in mutation avoidance, repeat tract instability, and flap cleavage.Mol. Cell. Biol. 21:4889–4899.
72. Yoon, J.-H., P. M. Swiderski, B. E. Kaplan, M. Takao, A. Yasui, B. Shen, andG. P. Pfeifer. 1999. Processing of UV damage in vitro by FEN-1 proteins aspart of an alternative DNA excision repair pathway. Biochemistry 38:4809–4817.
73. Zdraveski, Z. Z., J. A. Mello, M. G. Marinus, and J. M. Essigmann. 2000.Multiple pathways of recombination define cellular responses to cisplatin.Chem. Biol. 7:39–50.
6074 SPIRO AND McMURRAY MOL. CELL. BIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/m
cb o
n 05
Feb
ruar
y 20
22 b
y 17
5.20
6.37
.2.