sequence analysis – an overview a.krishnamachari [email protected]
TRANSCRIPT


Definition of Bioinformatics
• Systematic development and application of Computing and Computational solution techniques to biological data to investigate biological process and make novel observations

Research in BiologyResearch in Biology
OrganismFunctionsCellChromosomeDNASequences
General approach
Bioinformatics era

Information Explosion
• GENOME
• PROTEOME
• TRANSCRIPTOME
• METABOLOME

Databases
• Literature
• Sequences
• Structure
• Pathways
• Expression ratios

Databases
• Textual
• Symbolic (manipulation possible)
• Numeric (computation possible)
• Graphs (visualization )

January Issue

Integrated Database Search Engines
http://www.genome.ad.jp/dbget/
http://srs.ebi.ac.uk
http://www.ncbi.nlm.nih.gov/Entrez/


COG
Locus link
Uni Gene
Human – Mouse Map

Primary sequences
DNA Protein
StructuresExpression data
Pathways
Gene1000
Genome108

Analysis
• Individual sequences
• Between sequences
• Within a genome
• Between genomes

Sequence Analysis
• Sequence segments which has a functional role will show a bias in composition , correlation
• Computational methods tries to capture bias, regularities, correlations
• Scale invarient properties

Sequence Analysis
• Sequence comparison
• Pattern Finding –repeats, motifs,restriction sites
• Gene Prediction
• Phylogenetic analysis

TF
TF -> Transcription Factor Sites
TSS
TSS->Transcription Start Sites
RBS
RBS -> Ribosome Binding sites
CDS
CDS - > Coding Sequence (or) Gene
intergenic
-10-35

Protein-DNA interactions
• Biological functions
• Regulation or Modulation
• Specific binding (Specified DNA pattern)

DNA binding sites
• Promoter
• Splice site
• Ribosome binding site
• Transcription Factor sites
• Restriction Enzymes sites

The dimer is constructed such that it has bifold symmetry allowing the recognition helix of the second protein sub-unit to make the same groove binding interactions as the first. The distance between the recognition helices is 34 angstroms which corresponds to one turn of the B-DNA double helix. This means that when the recognition helix of one sub-unit binds in the groove of a specific region of DNA, the second sub-units' helix can also bind in the DNA groove, one turn along from the first helix

Odd
Even

DNA binding sites - Model
Experimental methods
Foot print expts. (Dnase )Methylation InterferenceImmuno precipitation assay
Compilation and Model building

TF1TF2TF3TF1TF1
-40-120-145
Design Oligos covering these regions for studying promoter activity
Carry out EMSA
Carry out Reporter assay
Carry out in-vivo experiments
Make Observations


Reporter GeneBS1BS2
-15-30-56-105
-150 -100 -50
Reporter Gene
Measure Expression
BS1
BS2 BS1

Statement of the problem
• Given a collection of known binding sites, develop a representation of those sites that can be used to search new sequences and reliably predict where additional binding sites occur.

Reference

1. Variability becomes inherent in biological sequences
2. manifesting at various length scales
3. Statistical and probabilistic framework is ideal for studying these characteristics

Sequence Analysis AND
Prediction Methods• Consensus• Position Weight Matrix (or) Profiles• Computational Methods
– Neural Networks– Markov Models– Support Vector Machines– Decision Tree– Optimization Methods

Strict consensus - TATA
Loose consensus - (A/T)R(G/C)YG
Weight matrix OR profile

Describing features using frequency matrices
Describing features using frequency matrices
• Goal: Describe a sequence feature (or motif) more quantitatively than possible using consensus sequences
• Need to describe how often particular bases are found in particular positions in a sequence feature

Describing features using frequency matrices
Describing features using frequency matrices
• Definition: For a feature of length m using an alphabet of n characters, a frequency matrix is an n by m matrix in which each element contains the frequency at which a given member of the alphabet is observed at a given position in an aligned set of sequences containing the feature

Frequency matrices (continued)Frequency matrices (continued)
• Three uses of frequency matrices– Describe a sequence feature– Calculate probability of occurrence of feature
in a random sequence– Calculate degree of match between a new
sequence and a feature

Frequency Matrices, PSSMs, and Profiles
Frequency Matrices, PSSMs, and Profiles
• A frequency matrix can be converted to a Position-Specific Scoring Matrix (PSSM) by converting frequencies to scores
• PSSMs also called Position Weight Matrixes (PWMs) or Profiles

Methods for converting frequency matrices to PSSMs
• Using log ratio of observed to expected
where m(j,i) is the frequency of character j observed at position i and f(j) is the overall frequency of character j (usually in some large set of sequences)
score(i) logm( j,i) / f ( j)

Finding occurrences of a sequence feature using a Profile• As with finding occurrences of a
consensus sequence, we consider all positions in the target sequence as candidate matches
• For each position, we calculate a score by “looking up” the value corresponding to the base at that position


Nucleotides
1 2 3 4 5
A x11 x21 x31 x41 x51
T x12 x22 x32 x42 x52
G x13 x23 x33 x43 x53
C x14 x24 x34 x44 x54
Positions (Columns in alignment)
TAGCT AGTGC x12 + x21 + x33 + x44 + x52
if is above a threshold it is a site
V1V1
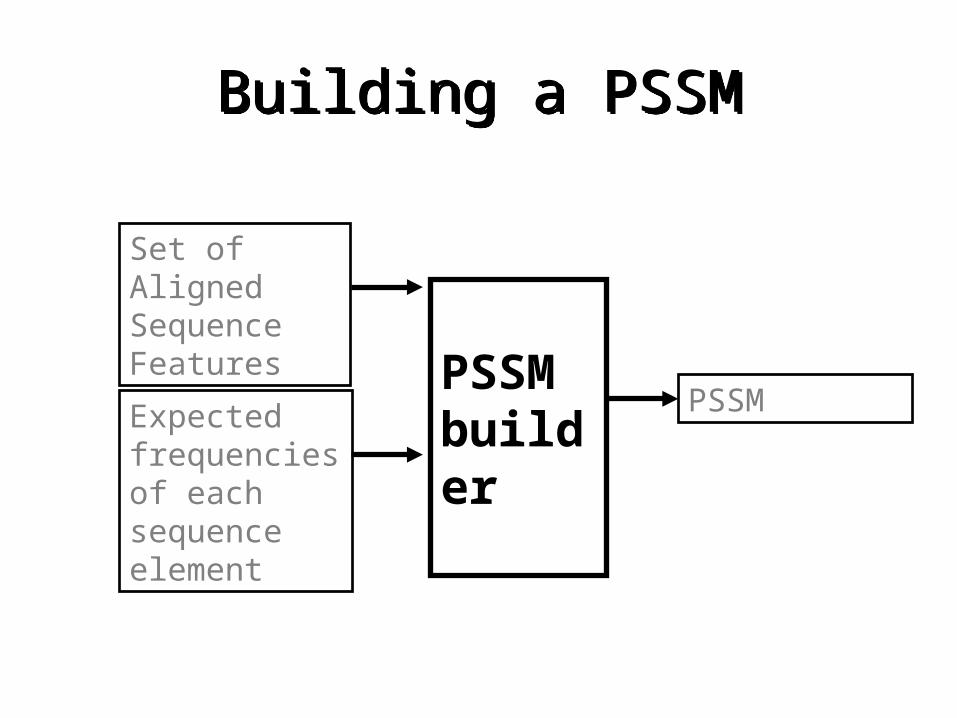
Building a PSSMBuilding a PSSM
PSSM builder
Set of Aligned Sequence Features
Expected frequencies of each sequence element
PSSM

Searching for sequences related to a family with a PSSM
Searching for sequences related to a family with a PSSM
PSSM search
PSSM
Set of Sequences to search
Sequences that match above threshold
Threshold
Positions and scores of matches
PSSM builder
Set of Aligned Sequence Features
Expected frequencies of each sequence element

Consensus sequences vs.
frequency matrices
Consensus sequences vs.
frequency matrices• consensus sequence or a frequency
matrix which one to use?– If all allowed characters at a given position
are equally "good", use IUB codes to create consensus sequence
• Example: Restriction enzyme recognition sites
– If some allowed characters are "better" than others, use frequency matrix
• Example: Promoter sequences

Consensus sequences vs.
frequency matrices
Consensus sequences vs.
frequency matrices• Advantages of consensus sequences:
smaller description, quicker comparison
• Disadvantage: lose quantitative information on preferences at certain locations

Shannon Entropy
• Expected variation per column can be calculated
• Low entropy means higher conservation
• Entropy yields amount of information per column

Entropy Or Uncertainty
• The entropy (H) for a column is:
• a: is a residue,
• fa: frequency of residue a in a column,
• fa Pa as N becomes large
)(
)log(aresidues
aa ffH
CGTAi
ii PPH,,,
log

Information
• Information Gain(I)= H before – H after
• H before =
CG,T,A,a
logH g aa ppGenomic composition
CG,T,A,i
iiafter p log p H

Information Content
• Maximum Uncertainty = log2 n
– For DNA, log2 4 = 2
– For Protein log2 20
Information content I(x)I (x) = Maximum Uncertainty – Observed Uncertainty
Note : Observed Uncertainty = Observed Uncertainty – small size sample correction
CGTAi
ii pp,,,
log2 I


Shine-Dalgarno Translation start site
Spacer

Binding site regions comprises of both signal(s)(binding site) and noise (background).
Studies have shown that the information content is above zero at the exact binding site and in the vicinity the it averages to zero
The important question is how to delineate thesignal or binding site from the background.One possible approach is to treat the bindingsite (signal) as an outlier from the surrounding(background) sequences.

Krishnamachari et al J.theor.biol 2004

Assumption of independence
• Prediction models assumes independence
• Markov models of higher order require large data sets
• This require better data mining approaches

Regulatory sequence analysis
• Analysis of upstream sequences of co-regulated genes (micro-array expts.)
• Phylogenetic foot-printing – Motif discovery
