sumario / contents - fcsd.orgºmero-1-marzo-2003_12278.pdf · los aspectos médicos del síndrome...
TRANSCRIPT

volumen 7 número 1 marzo 2003
• Editorial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
• Caracterización citogenética del síndrome de Downen la provincia de Camagüey . . . . . . . . . . . . . . . . . . . . . . . . . . . 2Cytogenetic characterization of Down syndrome in the Camagüey provinceH. PIMENTEL, E. DYCE
• Variante de Dandy-Walker asociado a síndrome de Down . . . . . . 6Dandy-Walker variant associated to Down syndromeJ. ROQUER
• Estudio de la situación actual y derechos constitucionalesde las personas con discapacidad . . . . . . . . . . . . . . . . . . . . . . . . 10Study on the actual situation and the constitutional rightsof the handicapped peopleA. VILÀ, M. TRUETA
• Congresos y reuniones . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
• Selección bibliográfica . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Sumario / Contents

Presidenta del Patronato de la FCSD:Montserrat Trueta
Director: Josep M. Corretger
Jefe de Redacción: Agustí Serés
Edición y coordinación: Katy Trias
Asesoría científica:Cardiología: J. CasaldáligaDermatología: J. FerrandoDietética-nutrición: A. GutiérrezEndocrinología: A. GodayEstomatología y Cirugía Máxilo-facial:A. MonnerGenética: A. SerésGinecología: J. CararachMedicina Interna: A. GarnachoNeurología infantil: R. GassióNeurología adultos: J. RoquerOdontopediatría: M.E. AlariOrtodoncia: M.A. MayoralOftalmología infantil: A. GalánOftalmología adultos: J.M. Simón, J. PuigOtorrino-laringología: J. DomènechPediatría: J.M. CorretgerPsicología: B. GarvíaPsiquiatría: J. BarbaTraumatología y Ortopedia: J.C. González
Consultores de medicina:F. Ballesta Martínez, M. Cruz Hernández, J.Moreno Hernando, C.J. Epstein (USA) y S.M.Pueschel (USA)
Consultores de psicopedagogía:Equipo de la FCSD: R. Borbonès, M.Casellas, A. Domènech, M. Golanó, J.M.Gracia, A. de Grecia, I. Jover, M.J. Miquel,M. Peralta, M. Roura, J. Ruf, J. Sanhueza, M.Torres, D. Torres y T. VallsT. VilàL. Brown (USA)
Edita:FUNDACIÓ CATALANA SÍNDROME DE DOWN (FCSD)Redacción:FCSDValència 229, pral. 08007 BarcelonaTeléfono: 93 215 74 23 - Fax: 93 215 76 99e-mail: [email protected]: http://www.fcsd.orgSecretaría de redacción: Joan C. Villalonga
El objetivo de SD. Revista Médica Internacional sobre el Síndrome de Down es, por un lado, recoger los conocimientos actuales sobrelos aspectos médicos del síndrome de Down y hacer una permanente revisión y actualización, desde los avances más prometedores enciencias básicas como la biología molecular y la genética, hasta la práctica clínica diaria; y, por otro lado, tratar aquellos aspectospsicopedagógicos que por su relación con el campo médico puedan tener un interés práctico para los pediatras generalistas y especialistasrelacionados con el síndrome. SD considerará para su publicación trabajos clínicos o de investigación relacionados con el síndrome deDown en todas sus ramas.
Este número ha sido posible gracias a la colaboración de LABORATORIOSURIACH.
© Fundació Catalana Síndrome de DownISSN: 1138-2074 D.L. B-40257-1986Imprime: Elite GraficPonts de Can Baruta, 2 , parc. 11- Molins de ReiExiste edición catalana de la revistaExiste versión digitalizada en inglés
NORMAS DE COLABORACIÓN
Trabajo original o de revisión
Trabajo de investigación, preferentementeprospectivo, sobre epidemiología, etiología, fisio-patología, anatomía patológica, clínica, y métodosde diagnóstico o terapéuticos. Los diseños reco-mendados son de tipo analítico, en forma de en-cuestas transversales, estudios de casos y contro-les, estudios de cohortes y ensayos controlados. Obien, trabajo de revisión o divulgación de aspectosdiversos. Extensión máxima: 25.000 espacios (re-súmenes incluidos), y un máximo de 10 tablas y/ofiguras. Se recomienda que el número de firman-tes no sea superior a 6 y el de referencias biblio-gráficas no supere las 20. Caso clínico. Extensión máxima: 8.000 espacios(resúmenes incluidos) y hasta 4 tablas y/o figuras.Número de firmantes no superior a 6 y el de re-ferencias bibliográficas no superior a 8.Avances psicopedagógicos. Aportaciones desdeel punto de vista educativo que, conjuntamentecon los aspectos médicos, favorezcan la calidadde vida de las personas Down y aporten una vi-sión antropológica de la persona. Extensión má-xima 12.000 espacios (resúmenes incluidos) yhasta 4 tablas y/o figuras. Número de firmantesno superior a 6 y el de referencias bibliográfi-cas no superior a 10.
Presentación y estructura de los trabajos
Se ajustarán a las recomendaciones del Co-mité Internacional de Editores de Revistas Médi-cas sobre los requisitos de uniformidad de los ma-nuscritos para revistas biomédicas (estilo Van-couver). Hojas DIN A4, mecanografiadas a dobleespacio y con márgenes suficientes, numeradascorrelativamente. Imprescindible también entre-gar una copia del trabajo en soporte magnético.
Se indicará: título del trabajo, nombre y pre-ferentemente primer apellido de los autores, si sondos, unidos por un guión; nombre y direccióncompleta del centro de trabajo; dirección para lacorrespondencia. Se hará constar expresamente siparte del trabajo ha sido objeto de presentación enalguna reunión o simposium o congreso, si hasido galardonado con algún premio y si ha reci-bido alguna subvención.Resumen. Los trabajos a publicar en las seccio-nes de trabajos originales o de revisión, casos clí-nicos y avances psicopedagógicos, tendrán que iracompañados de un resumen de un máximo de250 palabras en el idioma de presentación y eninglés, precedidos del título del trabajo en estosmismos idiomas. No contendrá datos que no seencuentren en el texto. Se indicarán hasta cincopalabras clave, de acuerdo con el Index Medicus.Redacción. Se recomienda la redacción en im-
personal. Conviene dividir claramente los traba-jos en apartados. Los originales en: Introducción,Material o Pacientes y Métodos, Resultados yDiscusión. Los casos clínicos en: Introducción,Observación clínica y Discusión. El texto de losavances psicopedagógicos, después del resumen,será desarrollado libremente por el autor.Agradecimientos. Se han de especificar breve-mente las contribuciones que requieren un agra-decimiento, especificando el tipo de ayuda –téc-nica o material– cuando la haya.Bibliografía. Las referencias, numeradas en eltexto por orden de aparición entre paréntesis, se-rán recogidas en una hoja aparte y siguiendo lasnormas de Vancouver.Tablas. Mecanografiadas en hojas independien-tes, y numeradas con cifras romanas. Han de sercitadas en el texto. Tendrán un título en la partesuperior. Si hay siglas, deben explicarse al pie dela tabla. Hay que evitar repeticiones entre tablas,figuras y texto.Figuras. Las indispensables para la buena com-prensión del texto. Se numerarán por orden de apa-rición con cifras arábigas. En una hoja aparte, semecanografiarán los pies. Las gráficas y dibujosdeben realizarse con impresión de alta calidad yentregarse en papel, indicando en el dorso el nom-bre del primer autor, y en la parte superior de la fi-gura su número. Si se utilizan fotografías de perso-nas, éstas no deben ser identificables, o deben iracompañadas por un permiso escrito para su utili-zación.Acrónimos, abreviaturas, símbolos y unidades.Es deseable utilizar el mínimo de acrónimos. Entodo caso se han de definir en su primera apari-ción. En las mediciones hematológicas y bioquí-micas se utilizará el sistema métrico de acuerdocon el sistema internacional de unidades (SI).
Entrega de los originales
Junto con una carta de presentación dondeconstará la aceptación de todos los firmantes, asícomo la certificación de la autoría y propiedad,se enviarán a la Secretaría de la FCSD.
Se acusará recibo de los originales y se infor-mará de su aceptación y fecha posible de publi-cación. Cuando el artículo se encuentre enprensa, el autor recibirá unas pruebas impresaspara corregir, que procurará devolver dentro delas 48 horas siguientes a su recepción.
Los editores podrán sugerir modificaciones enel texto cuando las crean necesarias, y tambiénrehusar la publicación de los trabajos que no con-sideren adecuados.
Indexada en EMBASE/Excerpta Medica e Índice Médico Español (IME)
La FCSD no se responsabiliza ni seidentifica necesariamente con las opi-niones emitidas por los autores de la re-vista.
SDREVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN
iii21
FUNDACIÓ CATALANA SÍNDROME DE DOWN

Síndrome de Down y salud mental
A menudo se especula sobre si las personas con síndromede Down (SD) son susceptibles de padecer enfermedadespsiquiátricas. En muchas ocasiones se ha supuesto que no su-frían estos trastornos –al menos no con la misma frecuenciaque la población en general– debido a su retraso mental.
Hay que ser prudente ante este tipo de afirmaciones, a ve-ces basadas en impresiones superficiales y estadísticas pocorigurosas. Cuando intervienen servicios de salud mental es-pecializados, adecuados y eficaces, y tanto las familias comolos profesionales aprenden a valorar su utilidad, la incidenciade las enfermedades mentales en estas personas demuestraser mayor de lo que imaginábamos.
Los familiares, maestros y demás profesionales que tene-mos a nuestro cuidado a personas con SD podemos incurriren el error de atribuir un trastorno del comportamiento o delcarácter, o incluso una disminución del rendimiento, al pro-pio retraso mental, sin tener en cuenta que puede tratarse deuna manifestación de un trastorno psiquiátrico superpuesto alretraso mental que, como tal, puede ser diagnosticado, tra-tado, e incluso curado.
En general, los estudios realizados al respecto parecen in-dicar que las personas con SD son menos vulnerables a lostrastornos mentales que aquellas que sufren un retraso mentaly no tienen SD. Sin embargo, de estas investigaciones tambiénse desprende que las personas con SD son más propensas a lademencia de Alzheimer y otras enfermedades neurodegene-rativas, así como a trastornos depresivos; en cambio, parecenestar algo más protegidas con respecto a la esquizofrenia, losestados paranoides y los trastornos de la personalidad.
Los trastornos emocionales y conductuales aparecen amenudo como complicaciones asociadas al retraso mental, ysuelen pasar inadvertidas o no reciben tratamiento. Las difi-cultades de las personas con SD para expresar verbalmentesus emociones comportan que, en su caso, determinadostrastornos se manifiesten de forma peculiar. Por ejemplo, ladepresión raramente es verbalizada y normalmente se mani-fiesta en forma de llanto, decaimiento general, aislamiento,pasividad y labilidad afectiva. De la misma forma, la sinto-matología de la ansiedad incluye trastornos del comporta-miento, irritabilidad, agresividad o tics como principales ma-nifestaciones.
Por otro lado, muchos trastornos depresivos y neuróticospresentan síntomas somáticos, con lo que se corre el riesgode no utilizar el tratamiento adecuado, y viceversa, ya quedeterminados síntomas psíquicos pueden ser la manifesta-ción de una enfermedad física. Así sucede en el hipotiroi-dismo, asociado con frecuencia al SD. Actualmente, el trata-miento de este trastorno ha dado lugar a una claradisminución de la sintomatología depresiva secundaria a laalteración tiroidea, cuyo diagnóstico era a menudo erróneo.
La disminución de la mortalidad y de la morbilidad al-canzada en las últimas décadas entre la población infantilcon SD, unida a una mayor predisposición a las enfermeda-
des neurodegenerativas, ha conducido al progresivo aumentode los trastornos psicogeriátricos en las personas con SD, enespecial de la enfermedad de Alzheimer, y se prevé que si-gan incrementándose, paralelamente al aumento de la espe-ranza de vida.
Esta cuestión añade complejidad a la relación de depen-dencia de la persona con SD con respecto a su familia, yaque la aparición de este tipo de enfermedades se suele pro-ducir cuando los padres ya han muerto o ya no pueden cola-borar –a causa de la edad– en la asistencia de la personaafectada.
En cuanto a los cambios socioculturales vertiginosos, losfenómenos de la inmigración y el desarraigo, la masificacióny despersonalización de las ciudades, el cambio en los mo-delos familiares y todos aquellos factores sociales que con-tribuyen al sufrimiento y a la enfermedad metal, dificultandoal mismo tiempo su asistencia y contención, tienen un efectoaún más negativo, si cabe, en las personas con SD que pade-cen un trastorno psiquiátrico. Éstas tropiezan con más difi-cultades para alcanzar una integración personal y una inser-ción social y laboral satisfactorias.
La calidad de la asistencia a personas con SD mejora díatras día, como demuestran una serie de pequeños detalles.Así, algo tan asumido como que su atención requiere la ver-dadera consideración de su individualidad –es decir, quesean tratados como sujetos, y no como objetos– hubiera po-dido juzgarse poco científico o fruto de la ingenuidad haceno muchos años. Hoy en día sabemos que, en un primer mo-mento, y para todo ser humano, el reconocimiento de unomismo implica el reconocimiento del otro, y que toda iden-tidad es única y valiosa. Sin embargo, también somos cons-cientes de que gran parte del funcionamiento del psiquismohumano, incluido el de la persona con SD, nos es todavíabastante desconocido.
En este sentido, la Fundació Catalana Síndrome de Down(FCSD) ha realizado un gran esfuerzo en los últimos añospara poder ofrecer a las personas con SD una atención inte-gral y pluridisciplinar, centrada en el tratamiento de las en-fermedades físicas y psíquicas, así como en las diversas ver-tientes de la socialización e integración laboral, con unaesmerada coordinación entre los profesionales que participanen este proceso.
Finalmente, sería necesario que las administracionescompetentes y los profesionales implicados avanzasen de co-mún acuerdo en lo que concierne a la adaptación de los dis-positivos asistenciales en salud mental a las necesidades delas personas con SD, que a menudo tienen dificultades pararecibir tratamiento a causa de la poca flexibilidad de las ins-tituciones.
En definitiva, tenemos que estar preparados para afrontartodos estos desafíos y problemas, y saber de antemano quesu solución dará lugar, sin duda, a nuevos retos. Esto, de hecho,significará que estamos avanzando...
JOSEP BARBA
Psiquiatra del Centro Médico Down. FCSD
SD2003: vol. 7, núm. 1 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 1
Editorial

SD2 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 2-5
Héctor Pimentel Benítez, Elisa Dyce GordonHospital Pediátrico Universitario Dr. Eduardo Agramonte Piña. Centro Provincial de Genética Médica. Camagüey, Cuba.
Correspondencia:Héctor Pimentel Benítez.Calle 2 # 6 e/ A y FinalRpto Las Mercedes. CamagüeyCP: 71200. Cuba.e-mail: [email protected]
Artículo recibido: 09.12.02
Resumen
Se presenta un estudio descriptivo transversalde caracterización citogenética a 143 pacientesdiagnosticados clínicamente como síndrome deDown en el Centro Provincial de Genética Mé-dica de Camagüey, en el período comprendidodesde 1986 hasta el 2000.
Los cromosomas se obtuvieron según procedi-miento seguido por Ram S. Verma y Harbin Babu(1995).
La trisomía libre por no disyunción cromosó-mica fue la principal causa de aparición de estesíndrome, seguido por el 9,09% de los casos conmosaicismo y un 4,86% correspondiente a lastranslocaciones robertsonianas; y entre estas lamás frecuente fue la t(14;21). Concluye con el comentario acerca de la importancia y vigenciaque tiene este tipo de estudio para el estableci-miento del diagnóstico, pronóstico y de la inte-gración social de las personas que tienen este sín-drome.
Palabras clave: síndrome Down, no disyunción,translocación, cromosoma 21, trisomía 21.
Cytogenetic characterization of Down syndromein the Camagüey Province
Abstract
A descriptive transverse study about the cyto-genetic characterization of 143 patients clinicallydiagnosed as Down syndrome was carried out atthe Camagüey Provincial Center of Medical Ge-netics, during the period 1986 to 2000.
Chromosomes were studied according toRam S. Verma and Harbin Babu method (1995).Free trisomy due to chromosomal non-disjunc-tion was the principal cause of this syndrome, fo-llowed by the cases of mosaicism (9,09%), and a4,86% were due to robertsonian translocation.Among translocations the more frequent was t(14;21). We conclude with the comment about theimportance and validity that this type of studyhave for the establishment of the diagnosis, prog-nosis and social integration of people with thissyndrome.
Key words: Down syndrome, non-disjunction,translocation, chromosome 21, trisomy 21.
Introducción
El síndrome de Down (SD), anomalía descritapor Langdon Down en 1866, constituye la princi-pal causa de retraso mental originado por anoma-lía cromosómica [1] y caracterizado clínicamentepor la presencia de una dismorfia facial y esque-
Original
Caracterización citogenética del síndrome de Downen la provincia de Camagüey

lética, así como por la presencia de malformacio-nes viscerales acompañando al retraso mental.
En Cuba tiene una incidencia de 9,8 x 10.000nacidos vivos [2], y en Camagüey nacen aproxi-madamente unos 10 niños con SD al año. El na-cimiento de un niño con este síndrome originatanto en sus padres como demás familiares graninquietud, dudas y angustia en nuestro medio.Sus padres se preocupan por el origen del pro-blema, las posibilidades de tratamiento y rehabi-litación, así como por el riesgo de recurrencia.Todo esto está muy relacionado con el meca-nismo de producción del síndrome, que puedepresentarse por el mecanismo de la no disyun-ción meiótica (como es el caso de la trisomía li-bre) que se presenta con más frecuencia en lasmujeres con edades avanzadas; debido a unatranslocación cromosómica (heredada o no); almosaicismo cromosómico o por la duplicaciónde una pequeña banda del cromosoma 21. De es-tos mecanismos depende en gran medida la pre-sentación clínica del síndrome, las posibilidadesde integración social, así como el riesgo de recu-rrencia o de repetición del mismo, por lo que sehace muy necesario la caracterización citogené-tica de cada niño o niña, independientemente deque su diagnóstico es eminentemente clínico.
Con el fin de caracterizar citogenéticamente alos niños y niñas con SD atendidos en la consultade genética clínica de la provincia de Camagüey serealizó este estudio que contribuirá a perfeccionarel asesoramiento genético a los familiares y orien-tar a los pediatras y dismorfólogos en cuanto al tra-tamiento médico y psicopedagógico de los niños yniñas; lo que garantizará mejorar el pronóstico, laintegración social y la prevención del síndrome.
Material y métodos
Se realizó un estudio descriptivo transversaldonde se recogen los resultados de los estudioscromosómicos en sangre periférica de 143 niñosy niñas diagnosticados clínica y citogenética-mente como SD, especificando su etiología, conel objetivo de caracterizar citogenéticamente a losmismos y exponer nuestra experiencia.
Para tal fin, se revisaron los libros de entradade muestras de sangre periférica al Laboratorio deCitogenética del Centro Provincial de GenéticaMédica de Camagüey, en el periodo 1986-2000.
La siembra de los linfocitos se realizó según la
técnica de microcultivo establecida por el CentroNacional de Genética Médica, utilizando mediode cultivo RPMI-1640.
Los cromosomas se cosecharon según el pro-cedimiento seguido por Ram S. Verma y HarbinBabu, en 1995 (protocolo 2.5) [3].
Se realizó bandeo cromosómico GTG, [4] y seanalizaron 20 metafases en cada caso. Se realizauna estadística descriptiva y se utilizan tablas dedistribuciones de frecuencias y figuras para facili-tar la interpretación de los resultados.
Resultados
Una serie de variantes cromosómicas dan ori-gen al SD, destacándose la trisomía libre por nodisyunción en un 86,01 % de los casos, seguidadel mosaicismo con un 9,09 % y las translocacio-nes robertsonianas con un 4,89 % (tabla 1). A dospacientes con el fenotipo del SD no se les demos-tró la presencia de anomalía cromosómica. La re-currencia del SD por trisomía libre se observó en3 casos (2,09%). Dos de ellos producto de lamisma pareja y el tercero era de otro matrimonio,siendo la madre la progenitora común. Tambiénse observó la ocurrencia del SD en tío y sobrinopor vía materna.
La tabla 2 muestra los resultados de las frecuen-cias de las translocaciones detectadas en esta serie,
SD2003: vol. 7, núm. 1, pp. 2-5 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 3
Tabla 1.Distribución de todos los casos de SD.
Tabla 2Distribución de los casos de SD
por translocación
Translocación Translocación(14;21) (21;21) Total
6 (85,7 %) 1 (14,3 %) 7 (100 %)
Trisomía Translo-libre cación Mosaico Total
123 (86,01 %) 7 (4,89 %) 13 (9,09 %) 143 (100 %)

donde se encontraron sólo dos tipos de ellas, lat(14;21) (Fig. 1) con un 85,7% y el 14,3% corres-pondió a la t(21;21). El 28,5% (No.=2) de estastranslocaciones eran heredadas. Un estudio fami-liar realizado en un caso de SD por t(14;21), de-tectó el origen paterno de la misma, la que tambiénse observó en una tía y abuela del “propositus”, de-mostrando así la segregación familiar (alterna) deldefecto (fig. 2), evitándose la propagación de sín-drome con el estudio citogenético del niño afec-tado y la caracterización citogenética de la familia.
Discusión
El cromosoma 21 es el autosoma más pequeño,
con apenas un 2% del total del genoma humano[5]; a pesar de ello, tiene gran significado clínico,y el hecho de que una gran parte del mismo estéconstituido por heterocromatina, hace que la triso-mía 21 sea bastante bien tolerada, pudiéndoseconstatar un rango de vida de más de 30 años enestas personas, llegando hasta los 50-60 años devida en un 25 % de ellos [6].
Los resultados mostrados revelan las causasmás frecuentes del SD. De acuerdo con los repor-tes de la literatura, la trisomía libre por no disyun-ción fue la causa mayormente presentada. En estoscasos, las personas presentan el fenotipo completodel síndrome y si ambos padres son cromosómica-mente normales son considerados como de bajoriesgo de repetición. En el caso de las transloca-ciones, clínicamente indistinguibles de los casosde trisomía libre, el riesgo de recurrencia dependede si los padres son portadores o no de la translo-cación y en este último caso el riesgo sería alto [7].En los casos de mosaicismo, el número o porcen-taje de células trisómicas y células normales, de-terminarán las manifestaciones clínicas y el pro-nóstico de la persona afectada. Es decir, a menornúmero de células trisómicas mejor pronóstico yviceversa. En la actualidad, se conoce que la du-plicación de los genes en la región crítica 21q22.3es la responsable del fenotipo SD y las manifesta-ciones clínicas también dependen del número degenes involucrados. La formación de isocromoso-mas pudiese ser otra causa, y en estos casos es indispensable realizar un estudio cromosómico fa-
SD4 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 2-5
Figura 1. Translocación robertsoniana t(14;21).
PADRE45, XY, t(14;21)
MADRE46, XX
Esperma Óvulo
HIJO
Síndrome Down46, XY, –14+ der (14;21)
Posibles segregaciones alternas
45, XY, t(14;21) 45, XX, t(14;21)
Síndrome Down
46, XY, –14+ der (14;21)
1 2
Figura 2. Estudio de segregación. Segregación alterna.

miliar con el objetivo de descartar una transloca-ción 21;21 en uno de los progenitores [8].
Resulta interesante la reaparición del SD en fa-milias donde las personas afectadas tienen fórmu-las cromosómicas correspondientes a una “no dis-yunción” o “trisomía libre” y, más aún, la partici-pación de la vía materna en los mismos.
Muchos autores han tratado de explicar las po-sibles causas de la no disyunción, entre las cualesse han descrito la edad materna avanzada, los ge-nes predisponentes, los efectos intercromosómi-cos, el mosaicismo parental, el envejecimientoacelerado en madres jóvenes y la presencia de an-ticuerpos antitiroideos maternos [9].
La no disyunción meiótica produce tambiénmultisomía, originando casos de aneuploidía do-bles, tal como se observó en un caso de esta seriedonde están involucrados el cromosoma 21 yun X, resultando un niño con fórmula cromosó-mica 48, XXY + 21.
Otro posible mecanismo de formación de fe-notipo SD es la duplicación de la banda 21q22,evidenciable únicamente por técnicas de biologíamolecular [10]. Una serie de recientes estudiosdelimitan la región crítica o región del SD, ya noa toda la banda 21q22, sino simplemente a lassubbandas 21q22.2 y 21q22.3, cuya presencia entriplicado provocará rasgos fenotípicos y defectoscaracterísticos del SD, lo que explica algunas va-riaciones interindividuales de ciertas personas.
Estas explicaciones y evidencias moleculares,permiten suponer, y de hecho asumir, que las mis-mas son los responsables de la aparición del feno-tipo SD en dos de los pacientes estudiados, losque citogenéticamente evidenciaron cariotiposnormales, suponiéndose entonces la presencia del“mosaicismo” no detectado en sangre periférica,motivo por el cual se les repitió el análisis cromo-sómico detallando un número mayor de metafa-ses, y mostrando nuevamente fórmulas cromosó-micas normales.
Si bien se plantea que la trisomía libre y latranslocación son clínicamente indistinguibles,también se conoce que en la duplicación el feno-tipo varía en dependencia de los genes que estánduplicados [6] y que los casos por mosaicismoproducen cuadros clínicos más leves y con mejorpronóstico. Estos conocimientos y los del riesgode recurrencia tienen gran importancia para elasesoramiento genético y la integración social delas personas con SD.
En conclusión, la caracterización citogenéticade las personas con SD tiene una trascendentalimportancia en la práctica médica y social, ya quela misma posibilitará el establecimiento de lacausa del SD, lo cual permitirá a su vez establecerel pronóstico y la evolución del mismo; de granvalor y a tener en cuenta para la integración socialde estas personas, como también será de gran in-terés para el asesoramiento genético y el cálculodel riesgo de recurrencia para las parejas deriesgo, así como para la población en general. Porlo que independientemente que el diagnóstico delSD sea eminentemente clínico, es necesario reali-zar este estudio a todas las personas afectadas.
Bibliografia
1. Thompson - Thompson. Genética médica. 4ed: 1996; 201-46
2. Ferrero Oteiza ME. Tendencias del Sín-drome de Down en Cuba. Su relación con laedad materna y tasa de fecundidad. Rev Cu-bana Pediatr 1998; 70: 141.
3. Verma RS, Babu A. Human chromosomes.Principles and techniques. 2 ed, New York1995; 9-29.
4. Catalogue for cell - culture, 1996-1997. LifeTechnologies Gibco – BRL, 1997; 92-3.
5. Capone GT. Down syndrome: Advances inMolecular Biology and the Neurosciences. JDev Behau Pediatr 2001; 22: 40-59.
6. Perera J. Introducción a la especificidad en elSíndrome de Down. Aspectos científicos.Masson, 1995; 58-60.
7. Viñas PC, Vega CV, Zaldivar VT, RodríguezGH, Lantigua CA. Síndrome de Down. Apropósito de 2 familias portadoras de translo-cación 14;21. Rev Cubana Pediatr 1999; 71:43-8.
8. Pimentel BH, Dyce GE, Dyce GB, Hallazgoscromosómicos en el laboratorio de citogené-tica del Hospital Pediátrico “Eduardo Agra-monte Piña”. Rev Electrónica. Arch Med Ca-maguey, 1995; 15-20.
9. RE Magenis. Invited editorial: On the originof chromosome anomaly. Am J hum Genet1988; 42: 529-33.
10. Petersen MB, Mikkelsen M. Nondisjunctionin trisomy 21: origin and mechanisms. Cy-togenet Cell Genet 2000; 91: 199-203.
SD2003: vol. 7, núm. 1, pp. 2-5 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 5

SD6 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 6-9
cial angioma and neurological malformation(Dandy Walker variant). The nosological situationof posterior fossa cyst malformations is reviewed,considering that the present case supports the uti-lity of the Dandy-Walker complex concept whichallows to place in the same syndromic group theposterior fossa malformations (megacisternamagna, Dandy-Walker syndrome, and Dandy-Walker variant)
Key words: Dandy-Walker Complex, Down syn-drome, cerebellar/vermian hypoplasia, ataxia.
Introducción
Los pacientes con síndrome de Down (SD)presentan con frecuencia malformaciones quepueden afectar diferentes órganos y sistemas [1].Son comunes las malformaciones congénitas car-díacas, las atresias digestivas (atresia duodenal y anal), las anomalías pulmonares (hipoplasiapulmonar), musculoesqueléticas y neurológicas.Dentro de las malformaciones neurológicas aso-ciadas al SD destacan el síndrome de Moya-Moya y la hipoplasia cerebelosa/vermiana. Pre-sentamos el caso de un paciente con SD asociadoa una malformación congénita de fosa posteriorque se correspondía con una variante de síndromede Dandy-Walker.
Caso clínico
Variante de Dandy-Walker asociadoa síndrome de Down
Jaume RoquerNeurólogo. Centre Mèdic Down. Barcelona.
Correspondencia:Dr. Jaume RoquerCentre Mèdic DownC/ València 229, pral, 1ª08007 Barcelona
Artículo recibido: 22.11.02
Resumen
Se presenta el caso de un paciente con sín-drome de Down y afectación multisistémica ca-racterizada por la presencia de una cardiopatíacongénita (canal auriculoventriclar común), an-gioma facial y malformación neurológica (va-riante de Dandy-Walker). Se revisa la situaciónnosológica de las malformaciones quísticas defosa posterior, considerando que el presente casoaporta datos en favor de la utilidad del conceptode complejo Dandy-Walker para situar sindrómi-camente en un mismo grupo a las malformacio-nes de fosa posterior (megacisterna magna, sín-drome de Danky-Walker y variante de Dan-dy-Walker).
Palabras clave: complejo Dandy-Walker, sín-drome de Down, hipoplasia cerebelo/vermiana,ataxia.
Dandy-Walker variant associated to Downsyndrome
Abstract
We report a patient with Down syndrome anda multisystemic affectation with congenital car-diovascular anomaly (atrioventricular canal), fa-
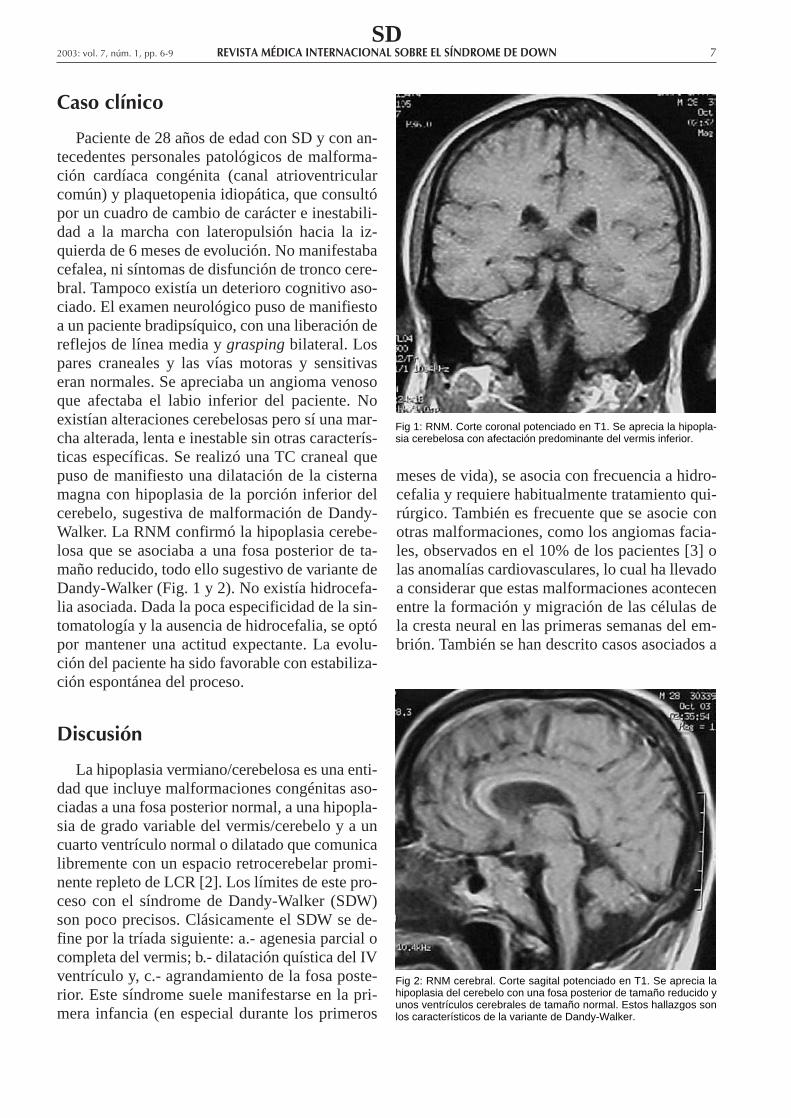
Caso clínico
Paciente de 28 años de edad con SD y con an-tecedentes personales patológicos de malforma-ción cardíaca congénita (canal atrioventricularcomún) y plaquetopenia idiopática, que consultópor un cuadro de cambio de carácter e inestabili-dad a la marcha con lateropulsión hacia la iz-quierda de 6 meses de evolución. No manifestabacefalea, ni síntomas de disfunción de tronco cere-bral. Tampoco existía un deterioro cognitivo aso-ciado. El examen neurológico puso de manifiestoa un paciente bradipsíquico, con una liberación dereflejos de línea media y grasping bilateral. Lospares craneales y las vías motoras y sensitivaseran normales. Se apreciaba un angioma venosoque afectaba el labio inferior del paciente. Noexistían alteraciones cerebelosas pero sí una mar-cha alterada, lenta e inestable sin otras caracterís-ticas específicas. Se realizó una TC craneal quepuso de manifiesto una dilatación de la cisternamagna con hipoplasia de la porción inferior delcerebelo, sugestiva de malformación de Dandy-Walker. La RNM confirmó la hipoplasia cerebe-losa que se asociaba a una fosa posterior de ta-maño reducido, todo ello sugestivo de variante deDandy-Walker (Fig. 1 y 2). No existía hidrocefa-lia asociada. Dada la poca especificidad de la sin-tomatología y la ausencia de hidrocefalia, se optópor mantener una actitud expectante. La evolu-ción del paciente ha sido favorable con estabiliza-ción espontánea del proceso.
Discusión
La hipoplasia vermiano/cerebelosa es una enti-dad que incluye malformaciones congénitas aso-ciadas a una fosa posterior normal, a una hipopla-sia de grado variable del vermis/cerebelo y a uncuarto ventrículo normal o dilatado que comunicalibremente con un espacio retrocerebelar promi-nente repleto de LCR [2]. Los límites de este pro-ceso con el síndrome de Dandy-Walker (SDW)son poco precisos. Clásicamente el SDW se de-fine por la tríada siguiente: a.- agenesia parcial ocompleta del vermis; b.- dilatación quística del IVventrículo y, c.- agrandamiento de la fosa poste-rior. Este síndrome suele manifestarse en la pri-mera infancia (en especial durante los primeros
meses de vida), se asocia con frecuencia a hidro-cefalia y requiere habitualmente tratamiento qui-rúrgico. También es frecuente que se asocie conotras malformaciones, como los angiomas facia-les, observados en el 10% de los pacientes [3] olas anomalías cardiovasculares, lo cual ha llevadoa considerar que estas malformaciones acontecenentre la formación y migración de las células dela cresta neural en las primeras semanas del em-brión. También se han descrito casos asociados a
SD2003: vol. 7, núm. 1, pp. 6-9 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 7
Fig 1: RNM. Corte coronal potenciado en T1. Se aprecia la hipopla-sia cerebelosa con afectación predominante del vermis inferior.
Fig 2: RNM cerebral. Corte sagital potenciado en T1. Se aprecia lahipoplasia del cerebelo con una fosa posterior de tamaño reducido yunos ventrículos cerebrales de tamaño normal. Estos hallazgos sonlos característicos de la variante de Dandy-Walker.

SD8 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 6-9
siringomielia [4] o a al síndrome de Coffin-Siris[5]. Los pacientes que, como el caso que se pre-senta, están afectados de una hipoplasia vermianaasociada a dilatación del IV ventrículo pero sinagrandamiento de la fosa posterior, se incluyendentro de la denominada variante de Dandy-Wal-ker. En la actualidad se considera que dentro delas anomalías quísticas de fosa posterior, tanto lamalformación de Dandy-Walker, como la va-riante Dandy-Walker y la megacisterna magnason expresiones diferentes de un mismo procesopor lo que desde ya hace años estas entidades seengloban dentro de un mismo término: el com-plejo Dandy-Walker [6].
Existen pocos casos descritos de asociaciónentre SD y de SDW [7], aunque la hipoplasia ce-rebelosa/vermiana, que puede considerarse comouna variante de SDW, es una malformación aso-ciada al SD con cierta frecuencia. En un intere-sante estudio que analizaba las alteraciones cro-mosómicas presentes en pacientes con diferentesgrados de agenesia vermiana [8], se comprobóque en los casos con agenesia vermiana inferior laexistencia de anormalidades cromosómicas, in-cluyendo el SD, eran más frecuentes (53%) queen los pacientes con agenesia vermiana completa(45%).
La sintomatología que el paciente presentaba,cambio de carácter e inestabilidad con lateropul-sión a la izquierda, sin otras anomalías en el exa-men neurológico no permite asegurar una rela-ción con la malformación cerebelosa, máximeteniendo en cuenta que el paciente parece excesi-vamente mayor para iniciar síntomas relaciona-dos con una malformación congénita. De todasformas tampoco puede descartarse completa-mente la relación, dado que existen casos deSDW de inicio en edad adulta [9] y, además, hayque recordar que la sintomatología atáxica quemanifestaba presenta una buena correlación clí-nico-radiológica con la zona del cerebelo hipo-plásica (vermis).
El tratamiento del SDW es, en general, quirúr-gico, en especial cuando se acompaña de hidroce-falia y existen signos de hipertensión intracraneal.Las posibilidades quirúrgicas varían desde la co-locación de un shunt ventriculoperitoneal, a la fe-nestración de la membrana quística, a la coloca-ción de un shunt primario quiste-peritoneal o conun shunt ventrículo-quiste-peritoneal [10]. En los
casos de variante de Dandy-Walker, la necesidadde intervención viene indicada por la presencia dehidrocefalia y de signos de hipertensión endocra-neal.
Finalmente, otro dato interesante es la afecta-ción multiorgánica del presente caso, que impli-caba al sistema nervioso central (variante deSDW), al sistema cardiovascular (canal atrioven-tricular común) y a la piel (angioma facial). Estasasociaciones están bien documentadas en los pa-cientes con SDW [3], lo que permite establecerun nexo entre ambos procesos e interpretarse portanto como un argumento más en favor de la si-militud nosológica entre el SDW y su variante.
Bibliografía
1. Kriss VM. Down syndrome: imaging of mul-tiorgan involvement. Clin Pediatr 1999; 38:441-9.
2. Kollias SS, Ball WS Jr, Prenger EC. Cysticmalformations of the posterior fossa: diffe-rential diagnosis clarified through embryolo-gic analysis. Radiographics 1993; 13: 1211-31.
3. Hirsch JF, Pierre-Kahn A, Renier D, Sainte-Rose C, Hoppe-Hirsch E. The Dandy-Walkermalformation. A review of 40 cases. J Neuro-surg 1984; 61: 515-22.
4. Hammond CJ, Chitnavis B, Penny CC,Strong AJ. Dandy-Walker complex and syrin-gomyelia in an adult: case report and discus-sion. Neurosurgery 2002; 50: 191-4.
5. Imai T, Hattori H, Miyazaki M, Higuchi Y,Adachi S, Nakahata. Dandy- Walker variantin Coffin-Siris syndrome. Am J Med Genet2001; 100: 152-5.
6. Barkovich AJ, Kjos BO, Norman D, EdwardsMS. Revised classification of posterior fossacysts and cystlike malformations based on theresults of multiplanar MR imaging. AJR AmJ Roentgenol 1989; 153: 1289-300.
7. Constantini S, Pomeranz S, Hoffman B, Mar-tin O, Rappaport ZH. Coexistence of Dandy-Walker syndrome and Down´s syndrome.Neurochirurgia 1989; 32: 56-7.
8. Chang MC, Russell SA, Callen PW, FillyRA, Goldstein RB. Sonographic detection ofinferior vermian agenesis in Dandy-Walker

malformations: prognostic implications. Ra-diology 1994; 193: 765-70.
9. Sato K, Kubota T, Nakamura Y. Adult onsetof the Dandy-Walker syndrome. Br J Neuro-surg 1996; 10: 109-12.
10. Kumar R, Jain MK, Chhabra DK. Dandy-Walker syndrome: different modalities of tre-atment and outcome in 2 cases. Childs NervSyst 2001; 17: 348-52.
SD2003: vol. 7, núm. 1, pp. 6-9 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 9
FUNDACIÓ CATALANA SÍNDROME DE DOWNC/ València, 229 - 08007 BARCELONA
Deseo recibir trimestralmente y de forma gratuita la revista SD-DS. REVISTA MÉDICA IN-TERNACIONAL SOBRE EL SÍNDROME DE DOWN. Remítanla, por favor, a la siguiente di-rección:
� CATALÁN � ESPAÑOL
Nombre: ............................................................................................................................................Domicilio: ........................................................................................................................................C. Postal: ............................ Población: ...........................................................................................
� Deseo, para colaborar con la FCSD, a partir del próximo número, recibir la revista porcorreo electrónico.
E-mail: ...............................................
Profesión: Firma:
Especialidad: Fecha:
Nota: Fotocopiar esta parte y enviarla por fax (932 157 699) o por correo ordinario a la FCSD. Gracias.
Póster publicado por la FCSD con motivo del Año Europeo de las personas con discapacidad.

SD10 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 10-16
Antoni VilàProfesor asociado de la Universitat de Girona y asesor jurídicode la Fundació Catalana Síndrome de Down.Montserrat TruetaPresidenta del Patronato de la Fundació Catalana Síndrome de Down.
Correspondencia:Sra. Montserrat TruetaFundació Catalana Síndrome de DownC/ València 229, pral. 08007 Barcelona
Artículo recibido: 25.10.02
Nota: El pasado 25 de septiembre de 2002 la Sra. Montserrat Trueta,Presidenta del Patronato de la Fundació Catalana Síndrome de Down, com-pareció en el Congreso de los Diputados a petición de la Subcomisión sobreel estudio de la situación actual de la discapacidad y perspectivas de futuro(154/011), constituida en el seno de la Comisión de Política Social y Em-pleo, para informar sobre la opinión de la Fundació Catalana Síndrome deDown referente al nuevo proyecto de ley para personas con discapacidad.
Resumen
Para analizar la situación actual, el artículo realizaun repaso histórico desde la creación de la Ley de In-tegración Social de los Minusválidos (LISMI), enca-minada a establecer una ley que regulara los dere-chos de las personas con discapacidad.
Después se hace un balance bastante exhaustivosobre su aplicación y el grado de desarrollo alcan-zado, que en términos generales se considera acepta-ble, aunque se identifican algunos aspectos mal re-sueltos y otros aún pendientes de desarrollar.
Finalmente, los autores concluyen citando las re-formas que hacen referencia al fondo, la estrategia ylos aspectos formales, así como las diferentes vías deactuación que, siguiendo el objetivo consensuado enla votación de la LISMI, considera esta ley como unpunto de partida para que las personas afectadas estu-vieran integradas en un plano de absoluta igualdad enla legislación general aplicable a todos los españoles.
Palabras clave: síndrome de Down, discapacidad,LISMI, legislación, vida independiente.
Study on the actual situation and the constitutionalrights of the handicapped people
Abstract
In order to analyse the current situation, the arti-cle gives an historical overlook since the approval ofthe law of Social Integration for Persons with Disa-bility (LISMI).
The purpose of the law was to regulate the rightsof persons with disability.
Following is a thorough study of the level of ap-plication which in general is considered acceptableeven though it points out some poorly resolved as-pects and others still awaiting development.
Finally, the authors mention the basis, the strategyused, the formal aspects and activities following theobjectives arrived at by consensus. The LISMI isconsidered a beginning as the intention is that handi-capped persons be integrated in the general legisla-tion applied to all Spaniards in absolute equalitywith the rest of the population.panyols.
Keywords: Down syndrome, disability, LISMI, le-gislation, independent life.
Ante todo quisiera agradecer la invitación queme han extendido para asistir hoy aquí para intentarcontribuir al estudio que tan oportunamente están
Avances psicopedagógicos
Estudio de la situación actual y derechos constitucionalesde las personas con discapacidad

llevando a cabo. Debo advertir, sin embargo, que nosoy persona de leyes. Mi única justificación es haberparticipado en el proceso de elaboración de laLISMI, filosofia que comparto totalmente, junto ami esposo Ramon Trias Fargas, y con el haber fun-dado la Fundació Catalana Síndrome de Down a raízdel nacimiento de nuestro hijo con síndrome deDown (SD).
La situación actual
Para tratar de la situación actual es convenientehacer una alusión, aunque muy breve, al nacimientode la LISMI, ley que ha marcado las políticas parapersonas con discapacidad en nuestro país en los úl-timos años.
Esta norma es fruto de la lucha de las personascon discapacidad, sus padres y amigos que en plenatransición a la democracia reivindicaron de formacontundente los legítimos derechos que les corres-pondían como personas. Estas reivindicaciones pro-piciaron la creación de la Comisión Especial delCongreso para el Estudio de los Problemas de losMinusválidos (1977) que fue presidida por mi es-poso Ramon Trias Fargas, entonces Diputado de lasCortes en representación de Convergencia Democrá-tica de Cataluña, y cuyos trabajos se dirigieron desdeel inicio al establecimiento, como ya habían hechootros países, de una ley que regulara los derechos delas personas con discapacidad.
Es singular la forma participativa que se estable-ció para elaborar los borradores de la norma, ya quese pretendía obtener un amplio y complejo consensode los afectados y sus familias, de los profesionales,así como de las fuerzas políticas. Este planteamientotenía el inconveniente de un largo y árduo proceso deelaboración (fue de más de cuatro años), pero lasventajas derivadas de un gran pacto superarían concreces dicha dificultad. La tenacidad de Ramon y lavoluntad de todos los sectores implicados consiguie-ron tejer un amplio y sólido acuerdo alrededor de laley, que se puso de relieve con su aprobación porunanimidad, ley que durante muchos años se deno-minó Ley Trias.
La filosofía que guió todo el proceso de elabora-ción de la ley y que enmarcó su contenido, se ba-saba en dos ejes, que seguramente es oportuno recor-dar en este análisis sobre la situación actual y la reflexión sobre el camino a emprender en el futuroque esta Subcomisión oportunamente ha empren-dido. El primero, lo señalaba con gran claridad elpropio Ramon con las siguientes palabras “... no en-tiendo esta Ley como algo perfecto, sino que, al con-trario, creo que se trata de un comienzo más que deun final. El objetivo a conseguir estriba precisa-
mente en llegar a la situación en que las leyes espe-ciales no sean necesarias para que las personasafectadas queden integradas en un plano de absolutaigualdad en la legislación general aplicable a todoslos españoles.”
Se pretendía, pues, hacer una ley especial paraeste colectivo con el fin de que nunca fuera necesariaotra porque las leyes ordinarias ya tenían en cuenta aestos ciudadanos. El segundo pilar de la ley estabaformado por los principios rectores de integración,normalidad y dignidad de las personas con discapa-cidad, supeditando a ellos todos los demás, incluidoslos aspectos económicos, como reflexionaba el ci-tado diputado y Presidente de la Comisión, “... yo nopuedo ignorar que el gasto público es siempre unacuestión de ordenar las preferencias sociales y paramí los destinatarios de esta Ley figuran muy arribaen la lista”.
Nuestro país dispone desde hace veinte años deuna Ley marco, con unos principios y postuladosacordes con los establecidos por las Naciones Unidasa nivel internacional, alrededor del Programa de Ac-ción Mundial para las Personas con Discapacidad.La vocación de globalidad de la ley obligó a regulartodos los ámbitos y facetas desde la misma preven-ción y el concepto de discapacidad, hasta los dere-chos a la asistencia sanitaria y farmacéutica, la reha-bilitación, la educación, el trabajo, los serviciossociales, la accesibilidad o las prestaciones económi-cas.
Después de dos décadas es sumamente oportunopreguntarse sobre la bondad de esta ley y de su apli-cación, ya que su carácter de ley marco exigía un dis-positivo reglamentario importante. Además, debe-mos interrogarnos sobre su validez actual. Sinperjuicio, de lo que se dirá más adelante, al hacer unbreve balance de su aplicación se considera que hacumplido, en buena medida, su misión: guiar la in-corporación de los aspectos relacionados con la dis-capacidad en la legislación ordinaria y que, por tanto,ha contribuido a hacer posible el futuro deseable queseñalaba Ramon.
Esta valoración general positiva está exenta decualquier triunfalismo, se es consciente de las insufi-ciencias de la ley y de algunos efectos perversos desu aplicación que luego se comentarán; así como, dellargo camino que todavía queda por recorrer hastaconseguir que las personas con discapacidad puedangozar de todos los derechos y deberes que les corres-ponden como ciudadanos. Se trata, pues, de escogerel camino adecuado y poner a disposición de los mis-mos los recursos necesarios para hacer efectivos di-chos derechos.
SD2003: vol. 7, núm. 1, pp. 10-16 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 11

• Balance de la aplicación de la LISMI y grado dedesarrollo alcanzado.
El Comité Asesor para la Atención y la Integra-ción de las Personas con Discapacidad de Cataluñaimpulsó la realización de un detallado Informe téc-nico sobre la aplicación de la LISMI en Cataluña, alos diez años de su aprobación. También el Real Pa-tronato sobre la Discapacidad y otras entidades efec-tuaron informes evaluativos semejantes de gran inte-rés. Quizá fuera necesario con motivo de los veinteaños de la LISMI realizar un estudio actualizadodesde el ámbito autonómico y estatal, en los ámbitosde sus respectivas competencias, con el fin de pro-fundizar en dichos análisis y poder detectar con ma-yor precisión los aspectos que merecerían modifica-ciones para alcanzar los objetivos propuestos.
En general las valoraciones efectuadas señalan unaceptable nivel de cumplimiento de la ley y la ten-dencia a incorporar a la normativa ordinaria muchosde los temas que antes eran objeto de normas espe-ciales; asimismo se identificaron algunos aspectosmal resueltos y otros pendientes de desarrollo. Acontinuación se efectúa un breve recorrido por loscapítulos de la norma señalando los aspectos que pa-recen más destacables.
a) Principios rectores. Se observa que con eltranscurso del tiempo se ha producido un de-bilitamiento de estos principios en algunossectores, como por ejemplo la integracióneducativa, la laboral o la normalización de al-gunos servicios sociales, como los residencia-les, todavía muy especializados, donde los po-deres de las entidades algunas veces pasan porencima de los intereses de la persona. En ge-neral se considera que deben hacerse mayoresesfuerzos normativos y económicos para al-canzar las cotas más altas posibles de vida au-tónoma e independiente, aspectos clave paraalcanzar la ciudadanía –en el sentido de sujetode derechos y deberes– de las personas condiscapacidad.
b) Prevención. No se llegaron a implantar losplanes generales que preveía la LISMI y a pe-sar que desde la Administración Central y lasComunidades Autónomas se han realizadoplanes y campañas, parecen insuficientes, es-pecialmente si tenemos en cuenta la alta si-niestralidad laboral y la cantidad de acciden-tes de circulación.
c) Diagnóstico y valoración. Éste es quizá unode los aspectos que exige una reflexión afondo, ya que existen una multiplicidad deequipos de valoración (seguridad social, servi-cios sociales, educación, etc.), muy burocrati-
zados y centrados en la discapacidad, en lugarde hacerlo en las capacidades y la orientaciónpara una vida autónoma.
d) Rehabilitación. El proceso rehabilitador se ha-lla todavía muy fragmentado y descoordinadoen distintas redes (sanidad, servicios sociales,educación, etc.) imposibilitando la rehabilita-ción integral de las personas con discapaci-dad. Por otra parte, para la realización de unavida autónoma es imprescindible un extensosuministro de prótesis, ortesis y ayudas técni-cas, que actualmente son insuficientes e in-completas.
e) Educación. El impacto de la LISMI en el sis-tema escolar ha sido considerable, como lomuestra el nivel de escolarización ordinaria.Sin embargo, todavía son bajos los niveles deintegración a partir de determinadas edades yniveles. Por otra parte, es notable la insufi-ciencia de recursos técnicos y humanos de so-porte necesarios para el éxito del proceso, unproceso, por otra parte, que se ha demostradotan beneficioso para el niño con discapacidadcomo para los compañeros de clase.
f) Trabajo. El acceso a un trabajo ordinario esclave en el proceso de integración social. Eldesarrollo de la LISMI ha posibilitado una ex-tensa red de centros especiales de empleo y decentros ocupacionales que deberían servir parala formación preparatoria a la integración y encambio sus cotas de integración en la empresaordinaria y en el empleo público son bajas. Lanormativa de ayudas y subvenciones priman elacceso al empleo protegido –actualmente agra-vada con las llamadas medidas alternativas alcumplimiento del 2%–, siendo ello un impor-tante elemento desintegrador. El cambio eneste apartado debería ser radical a favor de lacreación de puestos de trabajo normalizados enlas empresas, que deberían recibir incentivosclaros. En cuanto a la jubilación, la recientenormativa contempla el envejecimiento precozde las personas con discapacidad, y regula laposibilidad de jubilación anticipada, aunque lorelaciona con el porcentaje de discapacidad(65%) y no con el tipo de discapacidad, as-pecto que científicamente es más justificable,como por ejemplo la esperanza de vida de laspersonas con síndrome de Down, que segúnestudios recientes ha aumentado significativa-mente, pero aún son muy inferiores a la mediade la población. Si sólo se contempla a los quesuperan el 65%, quedan fuera un elevado nú-mero de personas que, aunque con menorgrado de discapacidad, también han trabajadoy deberían ser incluidos.
SD12 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 10-16

g) Prestaciones sociales y económicas. El sis-tema diseñado por la LISMI sufrió recortesimportantes, el primero debido a la interpreta-ción reglamentaria muy restrictiva que reque-ría un 65% de discapacidad e incluía a los fa-miliares en el cálculo de los ingresos, quedebió ser corregida por los tribunales. Des-pués sufrió una nueva regresión con las pen-siones no contributivas por invalidez que rein-corpora a los familiares que conviven con lapersona discapacitada para el cálculo del lí-mite de ingresos y, por otro lado, facilita el co-bro de la prestación por los padres a través delas prestaciones por hijos a cargo, en lugar deque la pensión la cobre directamente la per-sona afectada.
h) Servicios sociales. La red de atención prima-ria local se ha extendido en todo el país ofre-ciendo servicios generalizados de informa-ción, orientación, asesoramiento, entre otros.Sin embargo, la mayoría de los demás servi-cios obedecen a una tendencia marcadamenteespecializada, aspecto que colisiona con elprincipio de normalidad de la LISMI. Es ne-cesario que los servicios y equipamientossean lo más normalizados y flexibles posible.Para las personas con graves dificultades faltan programas de carácter sociosanitario.En los servicios sociales es donde se muestracon más claridad la falta de garantía de los de-rechos de las personas con discapacidad queles impide reclamar los servicios que pre-cisan.
i) Accesibilidad. En este punto la técnica y lasnormas jurídicas posteriores han superadoampliamente los planteamientos de la LISMI.Queda mucho por hacer, pero se ha avanzadoconsiderablemente en la accesibilidad al en-torno público, los edificios, el transporte y lacomunicación. Es necesario seguir en esteproceso emprendido desde la Administracióncentral, las CC.AA. y los ayuntamientos.
j) Financiación. Los presupuestos asignados adeterminados servicios y prestaciones son in-suficientes para garantizar los derechos de laspersonas con discapacidad y asegurar unavida autónoma, independiente y digna. Estainsuficiencia se hace especialmente visible,como se ha comentado, en los servicios socia-les.
k) Asociacionismo. El movimiento asociativo haaumentado y cambiado mucho en los últimosveinte años. Seguramente es necesario abrirun debate sereno y en profundidad sobre surepresentatividad, su vocación de interés pú-blico o su capacidad crítica. La ayuda desde
las administraciones es insuficiente y a me-nudo generadora de dependencia.
l) Competencias públicas. De la voluntad deglobalidad de la LISMI y su elaboración enplena transición resultó un marco competen-cial muy ambiguo. Están causando considera-ble confusión la poca claridad de la distribu-ción de las responsabilidades públicas, esdecir, entre el Estado, las CC.AA. y la admi-nistración local.
• Definir posibles vías de actuación de las refor-mas.
Parece que los próximos pasos a dar deben ser enla misma línea de la LISMI, incidiendo en accionesque contribuyan a hacer efectivos los derechos y re-formar aquellos aspectos de los que, según las valo-raciones efectuadas, se han constatado sus efectosnegativos. Para ello, se propone encargar a un grupode expertos a nivel estatal y de las Comunidades Au-tónomas que efectúen una evaluación detallada de laLISMI, su normativa de desarrollo y las normas or-dinarias que han incorporado las especificidades derivadas de la discapacidad, a partir de los análisisjurídicos y técnicos necesarios, así como de las opi-niones de los destinatarios de las normas y de las au-toridades responsables. En dicho informe deberíanidentificarse los puntos fuertes y débiles de este con-junto normativo y debería terminar con unas conclu-siones y con un conjunto de sugerencias y recomen-daciones.
A partir de dicho informe deberían tomarse deci-siones para la reforma. Una vía se referiría al fondo ysu estrategia debería dirigirse a garantizar los dere-chos de las personas con discapacidad y a exigir elcumplimiento de sus deberes. Este camino exige unexamen de las normas generales de los distintos ám-bitos, salud, educación, trabajo, servicios sociales,accesibilidad, etc. y establecer cuando sea menester,y sólo para lo necesario, las oportunas medidas dediscriminación positiva con el fin de facilitarles lasayudas o soportes precisos para garantizar la digni-dad que le corresponde como persona y conseguirque la libertad y la igualdad sean reales y efectivas,tal como propugna nuestra Constitución. La adop-ción de estas medidas exigen disponer de los equipa-mientos y sistemas lo más normalizados posiblesacordes con la discapacidad que presente la persona.
Una segunda vía se refiere a los aspectos forma-les, es decir, los referentes al modo de conseguirlo.Parece que el que mejor conjuga con los principiosde normalidad no es otra norma especial, sino la re-forma de las leyes ordinarias de los distintos secto-res, como la salud, la educación, el urbanismo o los
SD2003: vol. 7, núm. 1, pp. 10-16 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 13

servicios sociales para que incluyan en sus textos lasespecificidades que fuera menester para garantizarlos citados derechos. El responsable de la elabora-ción, aprobación y ejecución de dichas medidas se-rian los órganos legislativos y ejecutivos del Estado ode las CC.AA. según sus competencias.
• Propuestas de reformas a la legislación estatal.
Sin perjuicio de otras medidas que pudieran deri-varse de los estudios evaluativos citados, se consi-dera con carácter general que las principales refor-mas deberían referirse a los siguientes aspectos:
a) Concepto legal. Debería reconceptualizarsela discapacidad, de acuerdo con la últimaClasificación Internacional del Funciona-miento, de la Discapacidad y de la Salud(CIF) de la OMS, recientemente aprobadas yque ponen el énfasis en la importancia delentorno en la configuración de la discapaci-dad. Debería evitarse una declaración oficialde discapacidad por las consecuencias nega-tivas que tiene dicho etiquetaje. En este sen-tido la nueva definición de retraso mental dela Asociación Americana de Retraso Mentaldice: “El retraso mental no es algo que pue-des tener, como los ojos azules o el corazónenfermo. Está caracterizado por tu manera deser. No es un trastorno médico ni mental. Elretraso mental es una situación o forma defuncionar que se inicia durante la niñez y queestá caracterizado por una limitación, tantoen la inteligencia como en la habilidad deadaptación. El retraso mental refleja la con-tradicción existente entre las capacidades in-dividuales y el entorno.”
b) Principios. Además de los citados principiosrectores de la LISMI de integración y norma-lización, se propone profundizar dicha líneamediante la inclusión de la autonomía y la fi-losofía de vida independiente.
c) Diagnóstico y valoración. Es necesario des-burocratizar este acto, dándole un sentido dinámico y de orientación y asesoramiento sobre las capacidades residuales y las oportu-nidades que le brinda. Esta propuesta re-quiere cambios radicales en los equipos, susbaremos y criterios de actuación.
d) Prevención. Debe darse carácter prioritario ala prevención de las deficiencias, coordi-nando para ellos las investigaciones y accio-nes que se desarrollen en este ámbito.
e) Rehabilitación. Se propone una mejora de lacoordinación durante todo el proceso rehabi-
litador para darle un sentido integral, poten-ciando la red de equipamientos.
f) Educación-Formación. Deben emprendersemedidas decididas en las políticas de integra-ción de los niños con discapacidad tanto enlas primeras etapas de escolarización, comoespecialmente en la secundaria, la profesionaly la universitaria. Quizá fuera oportuno laelaboración de un libro blanco que evaluarala experiencia de estos años, aportara elemen-tos para la reflexión y fijara los principios deuna planificación integral de la integraciónescolar.
g) Trabajo. Se propone una política decidida eimaginativa de ocupación normalizada a tra-vés de medidas legales obligatorias y de fo-mento, asignando los medios económicospara hacerla efectiva. En este planteamientoel acceso al empleo público debería consti-tuir un ejemplo para las empresas. El trabajoprotegido y ocupacional debería ser lo másnormalizado posible y ser un puente haciael trabajo ordinario o destinado a las perso-nas que debido a su grave discapacidad lesfuera imposible el acceso a la empresa ordi-naria.
h) Vivienda. La problemática general de la vi-vienda se agrava en el caso de las personascon discapacidad. En este punto también seaboga decididamente por una política de nor-malización, facilitando a las personas condiscapacidad el acceso, la adaptación y finan-ciación permitiendo que sean ellas mismasquienes decidan con quién y dónde quierenvivir. Las viviendas tuteladas o residenciasdeberían tener también una función residualpara los discapacitados con graves problemasde dependencia y facilitar la emancipación delos que puedan.
i) Servicios sociales. Deberían potenciarse losservicios sociales destinados a posibilitar unavida autónoma e independiente. Se proponetambién con carácter urgente un proceso dedesinternamiento de las personas con disca-pacidad actualmente todavía en los hospitalespsiquiátricos.
j) Prestaciones económicas. Las pensiones de-berían garantizar una vida digna a las perso-nas con discapacidad y, para ello, es evidenteque las cuantías de las actuales pensiones nocontributivas son notablemente insuficientes;por otra parte, en la valoración de la situacióneconómica no deberían computarse los ingre-sos de la familia, sino únicamente la situacióndel solicitante que debe ser el receptor di-recto. Estas prestaciones básicas deberían
SD14 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 10-16

complementarse con otras de tipo ocasionalque cubrieran los gastos extraordinarios deri-vados de la discapacidad; sólo así, es posiblegarantizar la igualdad.
k) Accesibilidad. Debe seguirse el proceso em-prendido, intensificando las medidas técnicasy económicas para conseguir un entorno paratodos.
l) Coordinación. Debe mejorarse la coordina-ción, mediante mecanismos operativos y fle-xibles, tanto en el ámbito interno de las admi-nistraciones públicas como entre ellas y deéstas con el sector privado con el fin de opti-mizar los recursos. Para ello, podrían ser úti-les los mecanismos de planificación, a partirde unos mínimos consensuados entre los dis-tintos niveles territoriales.
m) Participación. Es necesario profundizar en laparticipación de los propios afectados, me-diante sistemas formales e incluso con otrasmedidas de innovación democrática. Se tratade que las personas con discapacidad ellasmismas y en su caso sus familiares puedanincidir en todas aquellas cuestiones que lesafectan, ya sean colectivas o individuales,tanto en el diagnóstico de la situación, comoen la toma de decisiones y la gestión de pro-yectos.
n) Iniciativa social. La LISMI reconocía el im-portante papel histórico desarrollado por lasentidades sociales de personas con discapaci-dad y las de sus familiares. Se considera quedebe seguir desarrollando este rol, garanti-zando la ley su autonomía e independencia,el respeto a su espíritu crítico y su lucha a fa-vor de los derechos legítimos de las personascon discapacidad. Las administraciones pú-blicas competentes deben facilitar el soportea dicho movimiento.
o) Investigación. Para avanzar en todas estasmaterias es necesario impulsar la investiga-ción en relación con los aspectos relaciona-dos con la discapacidad: salud, educación,ocupación, nuevas tecnologías y las ayudastécnicas.
Para terminar, si me permiten quisiera añadir unasreflexiones sobre mi experiencia personal con perso-nas con síndrome de Down.
En estos últimos 20 años, donde se ha desarro-llado la LISMI, aunque sea parcialmente, podemosencontrar, cada vez más, personas con SD que traba-jan en la empresa ordinaria con una identidad propiay un rol socio-laboral, y algunas personas que hanconseguido emanciparse y viven en su propia casa deforma prácticamente independiente.
Esto, aunque todavía desgraciadamente no es lanorma, hace 20 o incluso 10 años parecía imposible;sin embargo, es una realidad y demuestra que conapoyo a los padres, integración en todos los ámbitosde la vida y el necesario soporte a las necesidades deldesarrollo de la persona, todo es posible.
La LISMI fue un primer importante paso en reco-nocer y promocionar los derechos de las personascon discapacidad y a medida que se vayan adaptandoy aplicando las leyes veremos que estas personas, sitienen la oportunidad y ponemos sus intereses indivi-duales ante todo, nos sorprenden y nos hacen saberque todo vale la pena.
Ramon estaría muy contento si supiera que sucontribución ha dado fruto y que esta Subcomisiónelaborará un estudio que de seguro mejorará la situa-ción de las personas con discapacidad.
Por mi parte agradecer el esfuerzo de esta Subco-misión en nombre propio, el de mi hijo Andrés, y es-toy segura de que se sumarían al agradecimiento to-das las personas con discapacidad.
Bibliografía básica
Casado, D. Panorámica de la Discapacidad, Barce-lona, INTRESS, 1991.
Flórez, J. Aspectos orgánicos y clínicos del envejeci-miento y su incidencia en el comportamiento delas personas con síndrome de Down. En: Llegar aser adulto. Recopilación de las ponencias de lasVII Jornadas Internacionales sobre el Síndromede Down, Barcelona, Fundació Catalana Sín-drome de Down, 2000.
Generalitat de Catalunya, 10 anys de la Llei d’inte-gració social dels minusvàlids (LISMI) a Cata-lunya: present i futur. (2 vol). Barcelona, Depar-tament de Benestar Social, 1992 y 1993.
Naciones Unidas, Programa de Acción Mundial paralas Personas con Discapacidad y Normas unifor-mes sobre la igualdad de oportunidades para laspersonas con discapacidad, Madrid, Real Patro-nato de Prevención y de Atención a Personas conMinusvalía, 1996.
OMS Clasificación Internacional del Funciona-miento, de la Discapacidad y de la Salud, Madrid,IMSERSO, 2001.
Real Patronato de Prevención y de Atención a Perso-nas con Minusvalía. Desarrollo y aplicación de laLISMI (1982-1992), Madrid, Documentos n.16/92 (2 vol), 1992.
Real Patronato de Prevención y de Atención a Perso-nas con Minusvalía. Realizaciones sobre Discapa-cidad en España. Balance de 20 años. Madrid,1997.
SD2003: vol. 7, núm. 1, pp. 10-16 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 15

Trias Fargas, R. Presentación de la publicación de laLey de Integración Social de los Minusválidos,Barcelona, Generalitat de Catalunya, Departa-ment de Sanitat i Seguretat Social, 1982.
Vilà, A. Crónica de una lucha por la igualdad, Barce-lona, Fundació Institut Guttmann, 1994.
Vilà, A. Los derechos de las personas con dis-capacidad, en Servicios Sociales y Política So-cial, Madrid, Consejo General de Colegios Oficia-les de Diplomados en Trabajo Social, n. 43, 1998.
SD16 REVISTA MÉDICA INTERNACIONAL SOBRE EL SÍNDROME DE DOWN 2003: vol. 7, núm. 1, pp. 10-16
Fe de erratas
En el artículo de avances psicopedagógicos del an-terior número (Ruf J. El acceso al propio hogar: unanueva oportunidad para construir un proyecto de vida.SD-DS Revista Médica Internacional sobre el Sín-drome de Down 2002; 6: 43-6) la Figura 1 aparece demanera incompleta. La figura correcta es la que repro-ducimos a continuación:
Congresos y reuniones
II Rencontres Européennes sur la Trisomie 21“Trisomie 21 : quelle santé”
24, 25 y 26 de octubre de 2003Orléans, Francia
Información:F.A.I.T. 2110, rue du Monteil42000 – Saint-Etienne (França)Tel: 04.77.37.87.29Fax: 04.77.33.99.02E-mail: [email protected]
CAPACIDADESINTELECTUALES
CONDUCTAADAPTATIVA(conceptual, prácticay social)
PARTICIPACIÓN,INTERACCIONES YROLES SOCIALES
SALUD (salud, física,mental, etiología)
CONTEXTO SOCIAL(ambiente, cultura,oportunidades)
FUN
CIO
NA
MIE
NT
O
IND
IVID
UA
L
APOYOS
A.A.M.R. (2002)
Selección bibliográfica
LIBROS
Nutrición y dietética en la discapacidadManuel GallarElda, Asprodis, 2002, 262 pp. ISBN 84-607-5903-2
El estado nutricional óptimo contribuye a mejorar lacapacidad de respuesta del organismo frente a determi-nadas agresiones, además de facilitar el mantenimientode la salud y la prevención de enfermedades. Los cui-dados dirigidos a mejorar la situación nutricional delpaciente con discapacidad revierten directamente en sucalidad de vida. El libro «Nutrición y dietética en ladiscapacidad ofrece en primer término principios bási-cos en nutrición expuestos con precisión, claridad ysencillez, estableciendo normas racionales de alimenta-ción para las diferentes edades y las diversas situacio-nes patológicas consideradas. En segundo lugar des-cribe numerosas enfermedades y dificultades asocia-das, ofreciendo siempre soluciones prácticas en el tra-tamiento dietético y técnicas especiales de alimenta-ción.
Su valor práctico aumenta con la incorporación detablas que contienen datos de gran utilidad en la dieté-tica aplicada, así como herramientas necesarias parallegar a planificar el menú diario de un paciente condiscapacidad.
Por último, es necesario hacer hincapié en que el au-tor utiliza una pirámide de los alimentos no convencio-nal, pero que igualmente puede ser utilizada, siempresin olvidar una de las leyes fundamentales en nutriciónque es adaptar la alimentación a las necesidades indivi-duales.
Debe saludarse la aparición de este texto, dada la es-casez de publicaciones dedicadas específicamente altema que aborda en nuestro medio.
Alejandra Gutiérrez SánchezNutricionista-Dietista
Fundació Catalana Síndrome de Down
