the universal genetic test - los olivos women's medical
TRANSCRIPT

The UniversalGenetic Test
2200 Bridge Parkway, Suite 103 | Redwood City, CA 94065
www.counsyl.com | [email protected] | (888) COUNSYL

Counsyl Universal Genetic TestDisease BookABCC8-Related Hyperinsulinism ......................................................................................................................................................4Achromatopsia ................................................................................................................................................................................... 7Alkaptonuria ...................................................................................................................................................................................... 9Alpha-1 Antitrypsin Deficiency........................................................................................................................................................11Andermann Syndrome ......................................................................................................................................................................14ARSACS...........................................................................................................................................................................................16Aspartylglycosaminuria ....................................................................................................................................................................18Ataxia With Vitamin E Deficiency...................................................................................................................................................20Ataxia-Telangiectasia .......................................................................................................................................................................22Autosomal Recessive Polycystic Kidney Disease ........................................................................................................................... 24Bardet-Biedl Syndrome, BBS1-Related ...........................................................................................................................................26Bardet-Biedl Syndrome, BBS10-Related .........................................................................................................................................28Beta Thalassemia ..............................................................................................................................................................................30Biotinidase Deficiency......................................................................................................................................................................34Bloom Syndrome ..............................................................................................................................................................................36Canavan Disease .............................................................................................................................................................................. 38Carnitine Palmitoyltransferase IA Deficiency ..................................................................................................................................40Carnitine Palmitoyltransferase II Deficiency....................................................................................................................................42Cartilage-Hair Hypoplasia ............................................................................................................................................................... 45Choroideremia...................................................................................................................................................................................47CLN5-Related Neuronal Ceroid Lipofuscinosis.............................................................................................................................. 49Congenital Disorder of Glycosylation Type Ia .................................................................................................................................51Congenital Disorder of Glycosylation Type Ib.................................................................................................................................53Congenital Finnish Nephrosis.......................................................................................................................................................... 55Cystic Fibrosis ................................................................................................................................................................................. 57Cystinosis..........................................................................................................................................................................................62Factor V Leiden Thrombophilia .......................................................................................................................................................64Factor XI Deficiency.........................................................................................................................................................................67Familial Dysautonomia.................................................................................................................................................................... 69Familial Mediterranean Fever.......................................................................................................................................................... 71Fanconi Anemia Type C .................................................................................................................................................................. 73Fumarase Deficiency ....................................................................................................................................................................... 75Galactosemia.....................................................................................................................................................................................77Gaucher Disease................................................................................................................................................................................79GJB2-Related DFNB 1 Nonsyndromic Hearing Loss and Deafness............................................................................................... 83Glucose-6-Phosphate Dehydrogenase Deficiency........................................................................................................................... 85Glutaric Acidemia Type 1.................................................................................................................................................................88Glycogen Storage Disease Type Ia ...................................................................................................................................................91Glycogen Storage Disease Type Ib...................................................................................................................................................94Glycogen Storage Disease Type III ..................................................................................................................................................97Glycogen Storage Disease Type V ...................................................................................................................................................99GRACILE Syndrome......................................................................................................................................................................101Hereditary Fructose Intolerance......................................................................................................................................................103Hereditary Thymine-Uraciluria ......................................................................................................................................................105Herlitz Junctional Epidermolysis Bullosa, LAMA3-Related .........................................................................................................107Herlitz Junctional Epidermolysis Bullosa, LAMB3-Related..........................................................................................................109Herlitz Junctional Epidermolysis Bullosa, LAMC2-Related..........................................................................................................111
Page 2 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hexosaminidase A Deficiency........................................................................................................................................................113HFE-Associated Hereditary Hemochromatosis ............................................................................................................................. 116Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency ...........................................................................................120Hurler Syndrome.............................................................................................................................................................................123Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome ...........................................................................................126Hypophosphatasia, Autosomal Recessive ......................................................................................................................................128Inclusion Body Myopathy 2............................................................................................................................................................131Infantile Refsum Disease ................................................................................................................................................................133Isovaleric Acidemia ........................................................................................................................................................................135Krabbe Disease ...............................................................................................................................................................................137Leigh Syndrome, French-Canadian Type .......................................................................................................................................140Limb-Girdle Muscular Dystrophy Type 2E....................................................................................................................................142Long Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency .......................................................................................................144Maple Syrup Urine Disease Type 1B .............................................................................................................................................146Maple Syrup Urine Disease Type 3 ................................................................................................................................................149Medium Chain Acyl-CoA Dehydrogenase Deficiency ..................................................................................................................151Metachromatic Leukodystrophy .................................................................................................................................................... 153Mucolipidosis IV ............................................................................................................................................................................156Muscle-Eye-Brain Disease..............................................................................................................................................................158MYH-Associated Polyposis............................................................................................................................................................160Niemann-Pick Disease Type A.......................................................................................................................................................162Niemann-Pick Disease Type C ...................................................................................................................................................... 164Nijmegen Breakage Syndrome .......................................................................................................................................................166Northern Epilepsy ...........................................................................................................................................................................168Pendred Syndrome..........................................................................................................................................................................170Phenylalanine Hydroxylase Deficiency..........................................................................................................................................172Polyglandular Autoimmune Syndrome Type 1 ..............................................................................................................................176Pompe Disease ................................................................................................................................................................................179PPT1-Related Neuronal Ceroid Lipofuscinosis..............................................................................................................................181Primary Hyperoxaluria Type 1 .......................................................................................................................................................184Primary Hyperoxaluria Type 2 .......................................................................................................................................................186Pycnodysostosis ..............................................................................................................................................................................188Rhizomelic Chondrodysplasia Punctata Type 1 .............................................................................................................................190Salla Disease ...................................................................................................................................................................................192Segawa Syndrome...........................................................................................................................................................................194Short Chain Acyl-CoA Dehydrogenase Deficiency .......................................................................................................................196Sickle Cell Disease .........................................................................................................................................................................198Sjogren-Larsson Syndrome.............................................................................................................................................................202Smith-Lemli-Opitz Syndrome ........................................................................................................................................................204Spinal Muscular Atrophy................................................................................................................................................................206Sulfate Transporter-Related Osteochondrodysplasia..................................................................................................................... 209Tay-Sachs Disease ......................................................................................................................................................................... 212TPP1-Related Neuronal Ceroid Lipofuscinosis..............................................................................................................................214Tyrosinemia Type I.........................................................................................................................................................................217Usher Syndrome Type 1F ...............................................................................................................................................................219Usher Syndrome Type 3 .................................................................................................................................................................221Wilson Disease................................................................................................................................................................................223X-Linked Juvenile Retinoschisis ................................................................................................................................................... 225
Page 3 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
ABCC8-Related HyperinsulinismGene: ABCC8
Variant Accuracy Precision3992-9G>A 1.000 1.000F1388del 1.000 1.000V187D 1.000 1.000
Detection Rate Population<10% African American
87.60% Ashkenazi Jewish<10% Eastern Asia
42.90% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is ABCC8-related hyperinsulinism?ABCC8-related hyperinsulinism, also called congenital hyperinsulinism, is an inherited condition inwhich the pancreas releases inappropriately large quantities of the hormone insulin, leading to lowblood sugar (hypoglycemia). When blood sugar drops to dangerously low levels, seizures andpermanent brain damage may occur. If untreated, the condition could ultimately be fatal.
ABCC8 refers to the name of the gene that causes this disease. Other genes have been identified whichalso cause hyperinsulinism.
The pancreas normally secretes insulin in response to rising blood sugar. In people withABCC8-related hyperinsulinism, the pancreas secretes insulin even without sugar consumption,thereby removing too much sugar from the blood.
Infants with ABCC8-related hyperinsulinism tend to have significantly low blood sugar within the firstfew days of life. They often require immediate infusions of the sugar glucose to prevent seizures.These newborns are typically born larger than normal and may show difficulty feeding, poor muscletone, and breathing problems.
Page 4 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

In some people with ABCC8-related hyperinsulinism, symptoms do not appear until later in childhood.The low blood sugar associated with the condition can also range from mild to severe depending on theindividual, and varies even among members of the same family. Early and aggressive treatment isimportant to avoid permanent brain damage.
How Common is ABCC8-related hyperinsulinism?ABCC8-related hyperinsulinism affects roughly 1 in 50,000 Europeans. It is particularly commonamong people of Finnish and Saudi Arabian descent, where the disease may affect as many as 1 in2,500. A certain genetic mutation is prevalent in people of Ashkenazi Jewish descent.
How is ABCC8-related hyperinsulinism Treated?Treatments for ABCC8-related hyperinsulinism include dietary modification, medications, andsurgical intervention. The aim of treatment is to keep the affected person's blood sugar level in thenormal range to avoid brain damage.
If a child shows symptoms of ABCC8-related hyperinsulinism at birth, intravenous glucose is oftengiven to raise and stabilize the blood sugar level. Babies may need frequent feedings with largeamounts of carbohydrates, even overnight. A feeding tube may be helpful to ensure that a child getssufficient quantities of carbohydrates and may facilitate automatic feedings overnight.
There are several types of medication to treat ABCC8-related hyperinsulinism. These are typicallytaken orally and/or injected several times daily.
When diet and medication do not sufficiently manage blood sugar levels, the person may requiresurgery to remove part of the pancreas.
After an extended period of successful treatment, many people with ABCC8-related hyperinsulinismfind their symptoms lessen in severity or even go into remission.
People with ABCC8-related hyperinsulinism may find their symptoms aggravated by viral infectionsand should take particular precautions when they become ill, even if their symptoms have gone intoremission. They should also avoid long periods of time without eating.
What is the Prognosis for Someone With ABCC8-relatedhyperinsulinism?The long-term outlook for someone with ABCC8-related hyperinsulinism depends upon the severity ofthe symptoms and the vigilance of the efforts to treat it. Permanent brain damage can occur fromepisodes of low blood sugar. Even with treatment, people with the disease can develop some degree ofbrain damage or have learning difficulties. They also may be at an elevated risk of diabetes. In the
Page 5 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

most serious cases, when the disease is not recognized and properly treated, it can be fatal. Howeverwith careful treatment, people with ABCC8-related hyperinsulinism can live normal lifespans.
Page 6 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

AchromatopsiaGene: CNGB3
Variant Accuracy PrecisionR403Q 1.000 1.000E336X 1.000 1.000IVS8-3T>G 1.000 1.000819_826del8 1.000 1.000T383fs 1.000 1.000886-896del11insT 1.000 1.000
Detection Rate Population<10% African American
85.97% Ashkenazi Jewish<10% Eastern Asia
85.97% Finland85.97% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
85.97% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
85.97% Southern Europe
What is achromatopsia?Achromatopsia is an inherited disease that causes reduced visual acuity, an inability to see well inbright light, and the inability to see color.
The eyes have two types of photoreceptor cells: rod cells and cone cells. Cone cells function best inbright light and allow us to perceive color and fine detail. Rod cells, on the other hand, function best inlow light and are responsible for our night vision. Rod cells cannot perceive color and provide lessvisual acuity than cone cells.
In people with achromatopsia, cone cells do not function properly, leaving only rod cells. Because rodcells do not function well in bright light, as the amount of light increases, visual ability in people withachromatopsia decreases. People with achromatopsia will also be colorblind and will not perceivedetail well.
Most people with the disease have “complete achromatopsia,” meaning that none of their cone cells arefunctioning. Their vision is 20/200 or poorer and completely without color.
Page 7 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Some people with the disease have some functioning cone cells, leaving them with “incompleteachromatopsia.” They may be able to see some colors and typically have vision of 20/80 or poorer.Please note that Counsyl does not test for this form of the disease.
People with achromatopsia often experience a vibration or rapid oscillation in their field of vision, asymptom known as pendular nystagmus.
Symptoms of achromatopsia do not worsen over time and do not typically lead to blindness.
Several genes can cause achromatopsia. The gene for which Counsyl tests, CNGB3, is most commonlyreponsible for achromatopsia, causing 50% of known cases.
How Common is achromatopsia?Achromatopsia affects roughly 1 in 33,000 Americans. It is most common on the remote atoll ofPingelap in Pohnpei, part of the Federated States of Micronesia in the Western Pacific. There thedisease affects 4 to 10% of the population.
How is achromatopsia Treated?There is no cure for achromatopsia, but people with the disease have found ways to adapt. Manypeople with achromatopsia have found that dark brown, red, or gray-tinted glasses help them seeoutdoors during the day or in bright indoor spaces. Tinted contact lenses may also be helpful.
Other low vision aids such as large type books or magnifiers may be helpful. Parents of children withthe disease should work with their child’s school to make any necessary modifications to his or herlearning environment.
What is the Prognosis for Someone With achromatopsia?Achromatopsia does not affect lifespan, nor does it affect any other system of the body. While theperson will have poor eyesight, particularly in bright light, the disease is not progressive and will notlead to blindness.
Page 8 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

AlkaptonuriaGene: HGD
Variant Accuracy PrecisionG161R 1.000 1.000G270R 1.000 1.000P230S 1.000 1.000S47L 1.000 1.000M368V 1.000 1.000IVS1-1G>A 1.000 1.000IVS5+1G>A 1.000 1.000
Detection Rate Population<10% African American
84.38% Ashkenazi Jewish<10% Eastern Asia
84.38% Finland84.38% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
84.38% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
84.38% Southern Europe
What is alkaptonuria?Alkaptonuria is an inherited condition in which a particular enzyme does not function properly,creating a buildup of homogentisic acid (HGA) in the body. This accumulation causes alkaptonuria’smore visible symptoms, which include a brownish-black coloration to the urine when it mixes with airand a darkening of the body’s connective tissues after the age of 30. Excess HGA can also lead toarthritis, often beginning in early adulthood, as well as heart problems, kidney stones, and prostatestones.
A minority of patients do not have discolored urine, and for them arthritis may be the first noticeablesign of the disease. Arthritis often begins in the spine or major joints and appears in one’s 30s. In onelarge study of people with alkaptonuria, roughly half reported lower back pain in their 30s, and 94%reported the symptom by age 40. Hips, knees, and shoulders are frequently affected by alkaptonuria,though smaller joints are not. Studies indicate half of people with alkaptonuria require at least one jointreplacement by the age of 55.
Page 9 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

By the mid-60s, half of patients with alkaptonuria will have experienced kidney stones. Some menwith alkaptonuria also experience painful prostate stones. In their 60s, people with the diseasefrequently experience a hardening, thickening, and/or narrowing of the heart’s aortic or mitral valve.The coronary arteries of the heart may also harden.
Connective tissue pigmentation is most noticeable on the outside of the ear and may also be seen as apurple discoloration on the hands. The disease can also cause the body’s sweat to darken in color,which can stain clothing.
How Common is alkaptonuria?Alkaptonuria affects between 1 in 250,000 and 1 in 1,000,000 people globally. It is more common inthe Dominican Republic and in certain areas of Slovakia. In Slovakia, 1 in 19,000 people are affected.
How is alkaptonuria Treated?There is no treatment for the root cause of alkaptonuria. High doses of vitamin C have been shown todecrease the accumulation of pigment in the cartilage and may slow the development of arthritis.
Physical therapy can help patients maintain muscle strength and flexibility. Nearly all will requirelong-term pain management for their joint pain. Non-weight-bearing exercises, such as swimming,may be beneficial. Patients should avoid putting physical stress on their spine and major joints. For thisreason, heavy manual labor and high-impact sports are not recommended at any age.
Some patients will require surgery for joint replacement or kidney or prostate stone elimination.
What is the Prognosis for Someone With alkaptonuria?All people with alkaptonuria will experience chronic joint pain, usually beginning in their 30s. Thedisease does not reduce one’s lifespan.
Page 10 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Alpha-1 Antitrypsin DeficiencyIncluding Type Z Form and Type S Form
Gene: SERPINA1
Variant Accuracy PrecisionS allele 1.000 1.000Z allele 1.000 1.000
Detection Rate Population>99% African American>99% Ashkenazi Jewish>99% Eastern Asia>99% Finland>99% French Canadian or Cajun>99% Hispanic>99% Middle East>99% Native American>99% Northwestern Europe>99% Oceania>99% South Asia>99% Southeast Asia>99% Southern Europe
What is alpha-1 antitrypsin deficiency?Alpha-1 antitrypsin deficiency (AATD) is an inherited condition that can cause lung and liver disease.In some people, the disease may shorten lifespan.
The severity of AATD varies greatly from person to person—even among those in the same family.Knowing which mutations a child inherits can serve as some guide to how severe his or her symptomsmight be. In addition, smokers with the disease are much more likely to develop symptoms than non-smokers. Secondhand smoke, particularly from one's parents, can also increase the chances ofdeveloping symptoms.
As the name indicates, AATD is caused by a deficiency in a protein called alpha-1 antitrypsin. Thisprotein protects the body from an enzyme normally used to fight infection. Without sufficient alpha-1antitrypsin, an enzyme called neutrophil elastase, which normally fights infection in a helpful way, canattack and harm healthy tissue in the lungs. Abnormally formed alpha-1 antitrypsin can alsoaccumulate in and damage the liver.
People who inherit two copies of the Z allele (described in medical literature as "ZZ") are most likelyto have the severe symptoms of the disease, which are listed below. The Z allele causes the body toproduce very little of the alpha-1 antitrypsin protein. The S allele causes a moderately low level of
Page 11 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

alpha-1 antitrypsin to be produced, but it can often be enough to protect the body from lung damage.Those who have one Z allele and one S allele ("ZS") are more likely to have emphysema, particularlyif they smoke. People who have two S alleles ("SS") usually produce enough of the enzyme to avoidlung problems.
Emphysema, a chronic disease in which air sacs in the lungs lose their normal ability to expand andcontract, is the most common symptom of AATD. Emphysema causes a progressive difficulty inbreathing and a hacking cough. It can severely limit physical exertion. The first signs of emphysema,shortness of breath and wheezing, often appear between the ages of 40 and 50 in smokers with thedisease. Non-smokers with AATD typically develop emphysema symptoms later, even after the age of60.
Liver disease is another possible symptom of AATD. About 2% of children with AATD will developsevere liver complications. Common symptoms of these early liver problems include a swollenabdomen, swollen feet or legs, abnormal liver enzyme activity, and a yellowing of the skin or whites ofthe eyes (jaundice).
Overall, 15 to 19% of adults over the age of 50 with two Z alleles develop an accumulation of scartissue in the liver (cirrhosis). This symptom can develop at any age, with greater risk of cirrhosis laterin life. When liver disease associated with AATD begins later in life, destruction of the liver tissue canbe rapid.
Higher risk for a particular type of liver cancer has been reported among people with AATD, notablyin men.
Among carriers of AATD who have one copy of the Z allele and one normal gene, there is a slighlyelevated risk for lung or liver problems. One study placed this risk at 8%, versus 2 to 4% for thegeneral population. Smokers who are carriers of the Z allele are more likely to develop lung problems,while non-smoking carriers rarely do.
How Common is alpha-1 antitrypsin deficiency?In North America, AATD affects 1 in 5,000 to 7,000 people.
In a study of 75,000 Europeans, researchers estimated that 1 in 4,700 is affected by AATD. Thedisease is more common in Northern and Western Europe and less common in Eastern Europe. AATDaffects 1 in 1,500 to 3,000 Scandanavians. The disease is most common in Denmark, where it affects 1in 1,400, and least common in Russia, where it affects 1 in 86,000.
The Z allele is most common in northern Europe and lowest in Eastern Europe. The S allele, bycontrast, is most common in southern Europe and less so in northern Europe.
Page 12 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

AATD is rare in Asian and African populations, except in populations that are racially heterogeneous.For example, African-Americans in the United States have a higher rate of AATD than populations inAfrica.
Researchers believe that AATD is often diagnosed as chronic obstructive pulminary disease (COPD), arelatively common disease, without the realization that AATD is the cause of the COPD. For thisreason, the disease may be more common than prevalence numbers indicate.
How is alpha-1 antitrypsin deficiency Treated?It is strongly recommended that all people with AATD avoid smoking. Smokers are more likely todevelop symptoms of AATD. In smokers, symptoms tend to develop at an earlier age and progress at afaster rate. People with the disease should also avoid exposure to secondhand smoke, pollution,mineral dust, gas, and chemical fumes. Regular exercise and good nutrition are beneficial for peoplewith AATD.
Carriers of the Z allele should likewise avoid smoking. While most carriers of the Z allele will neverdevelop health problems related to it, smoking does increase the chance that symptoms will arise.
Infusions of purified human alpha-1 antitrypsin has been recommended for some people with AATD.It is considered most effective among people with moderate lung damage, though the overalleffectiveness of this therapy has not been adequately studied.
In people with severe liver or lung disease, transplantation of the failing organ may be an option. Lungtransplants have not been shown to improve lifespan, however. Liver transplants can “cure” the diseasebecause the donor liver will produce the alpha-1 antitrypsin protein.
What is the Prognosis for Someone With alpha-1antitrypsin deficiency?The severity of symptoms associated with AATD varies widely, making the prognosis equally varied.In some people, the disease can shorten lifespan while in others, it allows for a normal lifespan.Roughly 2% of children with two copies of the Z allele develop severe liver disease. Overall, smokersshow much more severe and rapid lung damage beginning earlier in life than non-smokers and thosewith one or more copies of the Z allele are more likely to develop symptoms than those with the Sallele. In non-smokers who develop lung complications after their 60th birthday, lifespan may benormal.
Page 13 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Andermann SyndromeGene: SLC12A6
Variant Accuracy PrecisionR675X 1.000 1.000Thr813fsX813 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland>99% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Andermann syndrome?Andermann syndrome, also called agenesis of corpus callosum with peripheral neuropathy, is aninherited disease causing progressive damage to the nervous system. Its symptoms appear early in lifeand include mental disability, a delay in motor skills, overall muscle weakness, curvature of the spine,and dysfunction in the nerves of the hands and feet resulting in numbness, pain, and muscle weakness.These symptoms will worsen over time.
The disease causes motor and sensory skills to be impaired from infancy. People with the disease alsoshare certain physical traits including a small head, long asymmetric face, small upper jaw, large ears,and a large distance between the eyes.
Two-thirds of people with the disease are missing the corpus callosum, a structure which connects theright and left sides of the brain, while the remaining third have a partially-formed corpus callosum.People with the disease learn to walk later than normal, often around the age of 3, and progressivelylose the ability to walk in their early teens. They may also experience seizures.
In their 20s, people with Andermann syndrome often develop hallucinations and psychosis. Thedisease is typically fatal before the age of 40.
Page 14 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The disease is seen almost exclusively in people from the Saguenay-Lac-St-Jean region of Québec,Canada.
How Common is Andermann syndrome?According to one researcher, Andermann syndrome affects 1 in 2,117 births in the Saguenay-Lac-St-Jean region of Québec, Canada, making 1 in 23 people there a carrier of the disease. It is rarely seen inany other population.
How is Andermann syndrome Treated?There is no cure for Andermann syndrome and few effective treatments for its symptoms. Physicaltherapy may be useful to maintain movement as long as possible. Surgery may also be recommendedto straighten the spine.
What is the Prognosis for Someone With Andermannsyndrome?Andermann syndrome is a progressive disease which impairs a person’s motor functions and causesmental disability. All people with the disease will eventually be wheelchair bound. In their 20s, peoplewith Andermann syndrome typically develop severe mental problems. The disease is usually fatalbefore the age of 40.
Page 15 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

ARSACSGene: SACS
Variant Accuracy Precision5254C>T 1.000 1.0006594delT 1.000 1.000
Detection Rate Population95.00% African American95.00% Ashkenazi Jewish95.00% Eastern Asia95.00% Finland95.00% French Canadian or Cajun95.00% Hispanic95.00% Middle East95.00% Native American95.00% Northwestern Europe95.00% Oceania95.00% South Asia95.00% Southeast Asia95.00% Southern Europe
What is ARSACS?ARSACS is the common name for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. It isa progressive disease that affects the body's ability to create a protein called sacsin, normally found inthe brain, skin, and muscles.
The first symptom, unsteady gait, typically appears between 12 and 18 months of age as toddlers beginto walk. Children also develop speech problems due to weak neck and facial muscles. The conditionbecomes increasingly worse over time, with muscle tension and spasms, difficulty coordinatingmovements, involuntary eye movements, and muscle wasting. Some people with ARSACS also losesensation in their arms and legs as the nerves degenerate.
Other symptoms include incontinence, deformities of the fingers and feet, and buildup of fatty tissueon the retina leading to vision problems. Occasionally, the disease also causes leaks in one of thevalves that control blood flow through the heart.
Most people with the condition are usually of normal intelligence and are able to live independentlywell into adulthood, although they eventually lose the ability to walk.
Page 16 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is ARSACS?The majority of people with ARSACS have ancestry in the Charlevoix-Saguenay region of Québec,Canada, where the condition affects approximately 1 in 1,500 to 2,000 people. Elsewhere in the world,the condition is rare.
How is ARSACS Treated?Treatment for ARSACS focuses on easing the symptoms and postponing major functional disabilities.Physical therapy and anti-spasmodic oral medications can help control muscle spasms, prevent jointand tendon deformities, and preserve muscle function for some time. Low doses of medication cancontrol incontinence. Occupational therapy and adaptive tools such as leg braces can support peoplewith ARSACS in daily tasks such as driving. As the disease progresses, however, people withARSACS typically lose the ability to perform these tasks. Children with the condition may benefitfrom speech therapy and other forms of support in school.
What is the Prognosis for Someone With ARSACS?People with ARSACS become wheelchair-bound at an average age of 41 and commonly die in theirfifties.
Page 17 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

AspartylglycosaminuriaGene: AGA
Variant Accuracy Precision199_200delGA 1.000 1.000C163S 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
99.00% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is aspartylglycosaminuria?Aspartylglycosaminuria (AGU) is an inherited condition in which an enzyme deficiency leads tovariety of physical symptoms. It is most common among people of Finnish descent.
Symptoms typically appear within the first few years of life. Infants with AGU experience frequentdiarrhea and infections. Other early signs include clumsiness, delayed speech, and hyperactivity.
People with the disease experience progressive mental disability, seizures, and behavioral problems.Between the ages of 13 and 16, they typically have the mental and motor development of a 5 or 6 year-old. By the mid-20s, they are severely mentally disabled.
People with AGU share certain physical features including sagging cheeks, eye deformities, a broadnose and face, a short neck, an asymmetrical head, and spinal deformities. Their facial features tend tocoarsen over time, and connective tissue problems or osteoporosis may develop.
How Common is aspartylglycosaminuria?Aspartylglycosaminuria is most common in Finland, where an estimated 1 in 26,000 babies areaffected. In some regions of Finland, where carrier rates can be 1 in 40, as many as 1 in 3,600 babieswill have the disease. AGU is the third most common cause of mental disability in Finland. Some
Page 18 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

studies have indicated that when the disease occurs in non-Finnish people, often the parents are closeblood relatives.
How is aspartylglycosaminuria Treated?There is no treatment for the cause of AGU. Medical professionals can only treat symptoms as theyarise. These treatments may include, but are not limited to, special education, anti-seizure medication,and orthopedic aids to help in movement.
What is the Prognosis for Someone Withaspartylglycosaminuria?The lifespan of a person with AGU has not been well-documented, perhaps due to the disease’s rarity.In one Canadian family, three affected siblings died in their 30s and 40s. All people with the diseaseexperience severe mental disability and impaired motor function.
Page 19 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Ataxia With Vitamin E DeficiencyGene: TTPA
Variant Accuracy Precision744delA 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic
87.50% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is ataxia with vitamin E deficiency?Ataxia with vitamin E deficiency (AVED) is an inherited disease that causes the nervous system todegenerate, leading to a progressive inability to control one’s voluntary movements (ataxia). If treatedearly and consistently with vitamin E, symptoms of the disease can be avoided.
If untreated with vitamin E, other symptoms of the disease can include difficulty speaking, loss ofsensation in the arms and legs, and loss of some visual acuity. In some people, intellectual decline andmental problems can occur. Other people with AVED have experienced heart problems as well.
In people with the disease who remain untreated, movement problems often begin between the ages of4 and 18 and worsen over time. Early symptoms often include clumsiness of the hands, problems withhandwriting, and reduced awareness of how one's body is positioned. These people will lose tendonreflexes in the arms and legs.
The type and severity of symptoms will vary from person to person, even among those in the samefamily.
How Common is ataxia with vitamin E deficiency?AVED is rare, but its exact prevalence is unknown. It may be more common in people ofMediterranean or North African descent.
Page 20 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is ataxia with vitamin E deficiency Treated?AVED is treatable with high doses of vitamin E taken regularly throughout life. If taken beforesymptoms begin, vitamin E can prevent symptoms from occurring at all. If symptoms have alreadybegun, vitamin E may prevent them from worsening and in some people, symptoms have beenreversed to some degree. Unsteadiness walking, however, often cannot be reversed.
People with AVED should not smoke, as this can reduce the amount of vitamin E in the body. Theyalso should not undertake jobs that require quick responses or good balance. Before learning to drive acar, their abilities should be assessed to determine whether driving is safe.
What is the Prognosis for Someone With ataxia withvitamin E deficiency?If treated with vitamin E before symptoms start, people with AVED can lead normal lives. Withouttreatment, people with AVED will become wheelchair-reliant between the ages of 11 and 50, and maydevelop significant physical and mental problems.
Page 21 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Ataxia-TelangiectasiaGene: ATM
Variant Accuracy PrecisionR35X 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is ataxia-telangiectasia?Ataxia-telangiectasia (A-T) is an inherited disease which affects a person’s ability to controlmovement. It also may weaken the immune system. People with A-T are at greatly increased risk forcancer, and the median age of death is around 22.
Shortly after children with A-T learn to walk, they will begin to wobble or stagger. Their motor skillswill develop slower than normal and they will have poor balance and slurred speech. They lose theability to follow objects with their eyes. This inability to control body movement, caused by damage topart of the brain, is called ataxia. By the age of seven or eight, children with the disease often lose themuscle control necessary to write, and most are confined to wheelchairs by the age of ten.
Teenagers and adults with the disease require help with everyday tasks, including dressing, eating,washing, and using the bathroom. Loss of muscle control often leads to drooling. While neurologicalproblems may impair their ability to communicate, people with A-T are usually of average or above-average intelligence.
Another hallmark of the disease is the appearance of tiny red spider-like veins around the corners ofthe eyes and on the ears and cheeks. This is known as telangiectasia.
Between 60 and 80% of people with A-T have weakened immune systems, leaving them prone toinfection, particularly in the lungs. They are also at an increased likelihood of developing cancer at an
Page 22 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

early age, particularly cancer of the blood (leukemia) and of the immune system (lymphoma). They arehypersensitive to the type of radiation found in X-rays and used in cancer treatment and typically mustavoid it.
Other symptoms of the disease may include diabetes, premature graying of the hair, problems withswallowing, and delayed sexual development.
A-T is caused by mutations in a gene involved in the control of cell growth and division, and also inthe repair of damaged DNA.
Carriers of A-T do not show symptoms of the disease, but studies have shown that they are at a greaterthan average risk of developing cancer, particularly breast cancer. They may also have an increasedrisk for heart disease.
How Common is ataxia-telangiectasia?A-T affects 1 in every 40,000 to 100,000 births worldwide. It is believed that around 1% of the U.S.population is a carrier of A-T.
How is ataxia-telangiectasia Treated?There is no cure for A-T, but symptoms of the disease can be addressed. Injections of gamma globulinmay be prescribed to help boost the immune system. High-dose vitamins may also be suggested.Antibiotics are typically used for infections. Vaccines for influenza and pneumonia may berecommended, as these diseases can be devastating to people with A-T.
Physical and occupational therapy are recommended to aid in movement and flexibility. Speechtherapy may also be useful.
What is the Prognosis for Someone With ataxia-telangiectasia?Nearly all people with A-T are wheelchair-bound by the age of 10. Because intelligence remainsnormal, many people with the disease graduate high school and college. People with A-T haveshortened lifespans, with the median age of death around 22 years. A small number of people havesurvived into their 40s and 50s. The most common causes of death from this disease are cancer, lunginfection, or lung failure.
Page 23 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Autosomal Recessive Polycystic KidneyDiseaseGene: PKHD1
Variant Accuracy Precision9689delA 1.000 1.000R496X 1.000 1.000V3471G 1.000 1.000Leu1965fs 1.000 1.000T36M 1.000 1.000
Detection Rate Population14.34% African American14.34% Ashkenazi Jewish14.34% Eastern Asia60.00% Finland14.34% French Canadian or Cajun14.34% Hispanic14.34% Middle East14.34% Native American14.34% Northwestern Europe14.34% Oceania14.34% South Asia14.34% Southeast Asia14.34% Southern Europe
What is autosomal recessive polycystic kidney disease?Autosomal recessive polycystic kidney disease (ARPKD) is an inherited disease in which clusters offluid-filled sacs (cysts) form in the kidneys, often leading to kidney failure by the age of 10 and areduced lifespan. According to studies, between 23 and 30% of infants with ARPKD die hours or daysafter birth due to breathing difficulties.
The majority of infants with ARPKD show enlarged, cyst-filled kidneys within the first month of life.These cysts will impair the kidneys’ ability to filter waste from the body. About 50% of infants withthe disease will also have an enlarged liver. These anomalies are often detectable through ultrasoundbefore the child is born. More than half of children will develop kidney failure by the age of 10.Without dialysis or transplantation, the disease is often fatal.
A minority of people with ARPKD do not show symptoms of the disease until later in childhood orearly in adulthood, with liver disease being the dominant symptom. In these people, the kidney diseaseis often mild.
Page 24 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Extremely high blood pressure is common in people with ARPKD. They are also prone to urinary tractinfections, frequent urination, low blood cell counts, pain in the back or the sides, varicose veins, andhemorrhoids. Many are also smaller than normal in stature.
How Common is autosomal recessive polycystic kidneydisease?ARPKD affects 1 in every 15,000 to 40,000 infants. However, the disease may actually be morecommon since people with milder forms of the disease may not be diagnosed without genetic testing.About 1 in 70 U.S. adults is thought to be a carrier of ARPKD.
How is autosomal recessive polycystic kidney diseaseTreated?The initial concern with infants who have ARPKD is to protect their ability to breathe. Eating anutritious diet can help the child’s growth, and in some cases, growth hormones are recommended.Infants and children may require feeding tubes in order to ensure proper growth.
If faced with kidney failure, people with ARPKD frequently undergo dialysis (a “cleansing” of theblood through a machine that remove wastes) or kidney transplants. If the liver is extremely damaged,transplantation of this organ may also be recommended. Some people with ARPKD must undergodialysis or a kidney transplant while they are still in infancy.
In all people with ARPKD, medications can lower blood pressure and clear up urinary tract infections.
What is the Prognosis for Someone With autosomalrecessive polycystic kidney disease?Between 20 and 30% of infants with ARPKD die hours or days after birth due to breathing difficulties.Of those who survive infancy, about 85% survive their first year of life, 82% survive to age 10, and73% live past the age of 15. In one study, 42% of those who survived their first year lived to the age of20.
As transplantation methods improve, it is expected that people with ARPKD will live longer lives.
Page 25 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bardet-Biedl Syndrome, BBS1-RelatedGene: BBS1
Variant Accuracy PrecisionM390R 1.000 1.000
Detection Rate Population80.00% African American80.00% Ashkenazi Jewish80.00% Eastern Asia80.00% Finland80.00% French Canadian or Cajun80.00% Hispanic80.00% Middle East80.00% Native American80.00% Northwestern Europe80.00% Oceania80.00% South Asia80.00% Southeast Asia80.00% Southern Europe
What is Bardet-Biedl syndrome, BBS1-related?Bardet-Biedl syndrome is an inherited disease that causes vision problems, kidney abnormalities,genital anomalies, extra fingers or toes, and mild obesity, among other symptoms. About half of peoplewith the disease have developmental delay or mental disability.
One hallmark of the disease is a vision problem caused by degeneration of the retina. It begins as nightblindness in childhood and progresses to a loss of peripheral vision. People with Bardet-Biedlsyndrome can also lose central vision during childhood or adolescence. The mean age at which theseadolescents become legally blind is 15.5 years. By early adulthood, they are severely visuallyimpaired.
Kidney abnormalities are present in most people with Bardet-Biedl syndrome. The problems caused bythese abnormalities can range from few functional problems to life-threatening kidney failure.
Around half of people with the disease have developmental disabilities. This can range from mildlearning disabilities or delayed emotional development to severe mental disability. In some cases thesedelays are due in part to vision loss, while in other cases they are a direct result of the disease.
Commonly, people with Bardet-Biedl syndrome have extra fingers and/or toes and mild obesity. Maleswith the disease often have small genitalia. Women with the disease typically have irregular menstrualcycles and may have structural deformities of the vagina. Some also have diabetes.
Page 26 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bardet-Biedl syndrome is similar to Laurence-Moon syndrome, and they have been thought to be oneand the same at times. The relationship between these two syndromes is still being studied.
How Common is Bardet-Biedl syndrome, BBS1-related?Bardet-Biedl syndrome is rare, affecting about 1 in 100,000 in North America and 1 in 125,000 inEurope. It is more or less common in specific populations, such as Kuwaiti Bedouins (1 in 13,500),residents of Newfoundland, Canada (1 in 17,500), and the Swiss (1 in 160,000).
How is Bardet-Biedl syndrome, BBS1-related Treated?There is no cure for Bardet-Biedl syndrome. Extra fingers and toes can often be surgically removed inchildhood. The vision and kidney problems associated with the disease can be treated in the standardfashion by medical specialists. If kidney problems reach life-threatening levels, dialysis and/or kidneytransplantation may be necessary. Diet and exercise can help control obesity. In women, vaginalmalformations can be surgically corrected.
What is the Prognosis for Someone With Bardet-Biedlsyndrome, BBS1-related?Kidney disease is a major cause of early death for people with Bardet-Biedl syndrome.
Page 27 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bardet-Biedl Syndrome, BBS10-RelatedGene: BBS10
Variant Accuracy PrecisionC91fs 1.000 1.000
Detection Rate Population46.00% African American46.00% Ashkenazi Jewish46.00% Eastern Asia46.00% Finland46.00% French Canadian or Cajun46.00% Hispanic46.00% Middle East46.00% Native American46.00% Northwestern Europe46.00% Oceania46.00% South Asia46.00% Southeast Asia46.00% Southern Europe
What is Bardet-Biedl syndrome, BBS10-related?Bardet-Biedl syndrome is an inherited disease that causes vision problems, kidney abnormalities,genital anomalies, extra fingers or toes, and mild obesity, among other symptoms. About half of peoplewith the disease have developmental delay or mental disability.
One hallmark of the disease is a vision problem caused by degeneration of the retina. It begins as nightblindness in childhood and progresses to a loss of peripheral vision. People with Bardet-Biedlsyndrome can also lose central vision during childhood or adolescence. The mean age at which theseadolescents become legally blind is 15.5 years. By early adulthood, they are severely visuallyimpaired.
Kidney abnormalities are present in most people with Bardet-Biedl syndrome. The problems caused bythese abnormalities can range from few functional problems to life-threatening kidney failure.
Around half of people with the disease have developmental disabilities. This can range from mildlearning disabilities or delayed emotional development to severe mental disability. In some cases thesedelays are due in part to vision loss, while in other cases they are a direct result of the disease.
Commonly, people with Bardet-Biedl syndrome have extra fingers and/or toes and mild obesity. Maleswith the disease often have small genitalia. Women with the disease typically have irregular menstrualcycles and may have structural deformities of the vagina. Some also have diabetes.
Page 28 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bardet-Biedl syndrome is similar to Laurence-Moon syndrome, and they have been thought to be oneand the same at times. The relationship between these two syndromes is still being studied.
How Common is Bardet-Biedl syndrome, BBS10-related?Bardet-Biedl syndrome is rare, affecting about 1 in 100,000 in North America and 1 in 125,000 inEurope. It is more or less common in specific populations, such as Kuwaiti Bedouins (1 in 13,500),residents of Newfoundland, Canada (1 in 17,500), and the Swiss (1 in 160,000).
How is Bardet-Biedl syndrome, BBS10-related Treated?There is no cure for Bardet-Biedl syndrome. Extra fingers and toes can often be surgically removed inchildhood. The vision and kidney problems associated with the disease can be treated in the standardfashion by medical specialists. If kidney problems reach life-threatening levels, dialysis and/or kidneytransplantation may be necessary. Diet and exercise can help control obesity. In women, vaginalmalformations can be surgically corrected.
What is the Prognosis for Someone With Bardet-Biedlsyndrome, BBS10-related?Kidney disease is a major cause of early death for people with Bardet-Biedl syndrome.
Page 29 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Beta ThalassemiaIncluding Beta Thalassemia Major and Beta Thalassemia Intermedia
Gene: HBB
Variant Accuracy PrecisionPoly A: AATAAA->AATGAA 1.000 1.000Poly A: AATAAA->AATAAG 1.000 1.000W15X 1.000 1.000Pro5fs 1.000 1.000Gly16fs 1.000 1.000Glu6fs 0.992 1.000Lys8fs 1.000 1.000Phe71fs 1.000 1.000IVS-II-705 1.000 1.000IVS-II-844 1.000 1.000Gly24 T>A 1.000 1.000-30T>A 1.000 1.000CAP+1 A>C 0.997 1.000-87C>G 1.000 1.000Hb C 1.000 1.000Hb E 1.000 1.000Hb O-Arab 1.000 1.000K17X 1.000 1.000Q39X 1.000 1.000619 bp deletion 1.000 1.000Phe41fs 1.000 1.000Ser9fs 1.000 1.000IVS-II-654 1.000 1.000IVS-II-745 1.000 1.000IVS-II-850 1.000 1.000IVS-I-6 1.000 1.000IVS-I-110 1.000 1.000IVS-I-5 1.000 1.000IVS-I-1(G>A) 1.000 1.000IVS-I-1(G>T) 1.000 1.000IVS-II-849(A>C) 1.000 1.000IVS-II-849(A>G) 1.000 1.000-88C>T 1.000 1.000-28A>G 1.000 1.000-29A>G 1.000 1.000
Page 30 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Detection Rate Population80.50% African American80.00% Ashkenazi Jewish80.00% Eastern Asia80.00% Finland80.00% French Canadian or Cajun80.00% Hispanic78.00% Middle East80.00% Native American80.00% Northwestern Europe80.00% Oceania90.00% South Asia81.00% Southeast Asia80.00% Southern Europe
What is beta thalassemia?Beta thalassemia is an inherited blood disease that affects hemoglobin, the major component of redblood cells which carry oxygen through the body. Hemoglobin is made up of two different oxygen-carrying proteins, alpha and beta. People with beta thalassemia do not produce enough betaprotein—and in some cases do not produce it at all—resulting in a shortage of red blood cells, acondition known as anemia. Without sufficient numbers of properly functioning red blood cells, theorgans of the body do not receive enough oxygen.
There are three main types of beta thalassemia. In the most severe form, thalassemia major (also calledCooley’s Anemia), a child will begin to show symptoms of severe anemia late in the first year of life.The lack of oxygen can cause him or her to be pale, listless, tired, and irritable. The child's spleen,liver, and heart may be enlarged, which is made noticeable by a swollen abdomen and yellowed skin.The child’s overall growth will be slowed and his or her bones may be thin, brittle, and/or deformed.Without frequent blood transfusions, the condition can be life-threatening at an early age.
Thalassemia intermedia, a less severe form of the condition, causes mild to moderate anemia and awide spectrum of possible health problems. The types of symptoms are the same as with thalassemiamajor, including bone deformities and an enlarged spleen, though these are typically not as severe.Thalassemia intermedia may not be diagnosed until later in life. People with thalassemia intermediarequire fewer blood transfusions and use them to improve the quality of their lives.
Sickle cell disease, which is described separately, is a type of beta thalassemia caused by the Hb Smutation. Sickle cell disease is caused by two copies of the Hb S mutation, or one copy of Hb S alongwith a beta thalassemia mutation. A carrier of a beta thalassemia mutation whose partner is a carrier ofthe Hb S mutation is at risk for having a child with sickle cell disease.
Page 31 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is beta thalassemia?Beta thalassemia is considered common worldwide, though its exact frequency is unknown. Thedisease is most common in people of Mediterranean descent, especially in those from Sardinia andCyprus. In Cyprus, 1 in 7 people are carriers of the disease, a rate which prompted a successfulgovernment-run prevention program there. Beta thalassemia is also commonly found in the MiddleEast and Asia.
Ethnic Group Carrier Rate Affected Rate
Cypriot 1 in 7 1 in 170
Sardinian 1 in 8 1 in 240
Italian 1 in 31 1 in 3,700
Middle Eastern 1 in 34 1 in 4,500
Southeast Asian 1 in 35 1 in 4,800
East Asian 1 in 62 1 in 15,000
Indian 1 in 64 1 in 16,000
How is beta thalassemia Treated?The most common treatment of beta thalassemia is blood transfusions, which provide a temporarysupply of healthy red blood cells to bring oxygen to the body. Among people with thalassemia major,transfusions may take place every two to three weeks. While these transfusions can be life-saving andlife-enhancing, they result in a toxic buildup of iron in the blood. To counteract this side-effect, peoplewith beta thalassemia require a procedure called chelation therapy in which a medication is taken toeliminate excess iron from the body. These individuals require frequent monitoring by a physician toassess the efficacy of transfusion/chelation therapy.
In a small minority of people, a bone marrow transplant from a sibling or other suitable donor has beenable to cure the disease. This procedure, however, is risky and could even be fatal.
What is the Prognosis for Someone With betathalassemia?Without proper diagnosis and treatment, people with beta thalassemia major have stunted growth andshortened lifespans. With proper treatment, growth may be normal and the person’s lifespan may
Page 32 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

extend into his or her 30s, 40s, and 50s. People with thalassemia intermedia who have less extremesymptoms may live a normal lifespan.
Page 33 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Biotinidase DeficiencyGene: BTD
Variant Accuracy PrecisionG98:d7i3 1.000 1.000A171T 1.000 1.000D252G 1.000 1.000F403V 1.000 1.000Q456H 1.000 1.000R538C 1.000 1.000D444H 1.000 1.000
Detection Rate Population89.02% African American89.02% Ashkenazi Jewish89.02% Eastern Asia89.02% Finland89.02% French Canadian or Cajun89.02% Hispanic89.02% Middle East89.02% Native American89.02% Northwestern Europe89.02% Oceania89.02% South Asia89.02% Southeast Asia89.02% Southern Europe
What is biotinidase deficiency?Biotinidase deficiency is a highly-treatable inherited disease in which the body cannot process thevitamin biotin due to a deficiency in a particular enzyme. If left untreated, the disease can causenumerous life-threatening complications. By taking daily supplements of biotin before symptomsoccur, however, all symptoms of the disease can be avoided. With early detection and treatment, aperson with biotinidase deficiency can live a completely normal life.
If the condition is not detected early and promptly treated with biotin, people with biotinidasedeficiency can experience seizures, poor muscle tone, difficulty with movement and balance, visionand/or hearing loss, skin rashes, breathing problems, fungal infections, and delayed mentaldevelopment. These symptoms often begin after the first few weeks or months of life and can be life-threatening if untreated.
If symptoms have already appeared, treatment with biotin can reverse damage to the body already doneby the disease. Vision or hearing loss and developmental delay, however, are irreversible.
Page 34 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

People who have less than 10% of the normal amount of the enzyme biotinidase are said to haveprofound biotinidase deficiency. Without treatment, their symptoms tend to be significant. People whohave between 10 and 30% of the normal amounts of biotinidase have a milder form of the diseaseknown as partial biotinidase deficiency. They may experience less severe symptoms, or may beasymptomatic until periods of illness or stress. Both forms of the condition can be successfully treatedwith biotin.
How Common is biotinidase deficiency?Overall, 1 in 60,000 births will be affected by either profound or partial biotinidase deficiency.Profound biotinidase deficiency, which is the most severe form of the disease, occurs in about 1 in137,000 births while the milder partial biotinidase deficiency occurs in about 1 in 110,000 people. Inthe general population, 1 in 120 people are carriers for biotinidase deficiency.
How is biotinidase deficiency Treated?Biotinidase deficiency is treated with a biotin pill taken daily by mouth. A physician can determine theproper dosage and adjust that dosage over time if necessary. This treatment is lifelong and highlyeffective. Biotin is non-toxic, so it is recommended that people with partial biotin deficiency also takebiotin supplements.
If treatment is begun after symptoms appear, some symptoms, such as skin problems and hair loss, willdisappear. If the disease has already caused irreversible hearing or vision loss, low vision aids orhearing aids may be helpful. Learning specialists can assist with any irreversible developmentaldeficits.
What is the Prognosis for Someone With biotinidasedeficiency?With early diagnosis and treatment, people with biotinidase deficiency can live completely normallives with no symptoms. Those in whom the disease is not detected early may experience permanentdamage to their hearing, vision, or intellect. In cases where the disease is entirely unrecognized, it canbe life-threatening.
Page 35 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bloom SyndromeGene: BLM
Variant Accuracy Precision2281del6ins7 1.000 1.0002407insT 1.000 1.000
Detection Rate Population<10% African American
97.00% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Bloom syndrome?Bloom syndrome, also known as Bloom's syndrome, is an inherited condition most easily noticed bythe person's extremely small stature. People with Bloom syndrome have chromosomes which oftenbreak and re-arrange. This instability results in high rates of cancer early in life. Some people with thedisease develop cancerous tumors before the age of 10, but more commonly cancer appears beginningin the late teens or early to mid-20s.
People with Bloom syndrome have a high-pitched voice and distinct facial features including a long,narrow face, small lower jaw, prominent nose and ears, and red lesions on the cheeks and the bridge ofthe nose (often described as “butterfly-shaped”) which appear and worsen with sun exposure.
Many, though not all, people with Bloom syndrome have learning disabilities or mental disability.They may also have diabetes, chronic lung problems, and suppressed immune systems that leave themunable to ward off infection as easily as most people. They tend to have high rates of pneumonia andear infections.
Men with Bloom syndrome are usually infertile. Some women with the disease have given birth tohealthy children. Women often experience early menopause.
Page 36 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Bloom syndrome?Bloom syndrome is very rare, although its frequency is unknown. Approximately one-third of peoplewith the disease are of Ashkenazi Jewish descent, making it more common in this population than inothers. Roughly 1 in 48,000 Ashkenazi Jews is affected by the disease.
How is Bloom syndrome Treated?There is no cure for Bloom syndrome. Children with Bloom syndrome need nutritional monitoring toensure maximum growth. Experiments with growth hormones in Bloom patients have been largelyunsuccessful. People with the disease are advised to stay out of the sun and wear sunscreen,particularly during childhood, to prevent skin lesions. They should also make an effort to avoidinfection of all kinds. In school, they may require special education classes due to learning difficulties.
People with Bloom syndrome are prone to cancer, so they should be screened regularly starting inchildhood and with increasing vigilance into adulthood. Because they are particularly sensitive toradiation and DNA-damaging chemicals, standard cancer treatments often need to be modified. Ifdiabetes is present, this condition is typically treated with diet, blood sugar monitoring, and insulinsupplements.
What is the Prognosis for Someone With Bloomsyndrome?Despite dealing with numerous medical problems, people with Bloom syndrome can lead productivelives. They are of normal or near-normal intelligence. While men with Bloom syndrome are infertile,some women have given birth to healthy children. Typically people with Bloom syndrome leadshortened lives, although lifespan can vary greatly from person to person. The cause of death is usuallycancer, which can occur in childhood, but more commonly appears in the late teens or early to mid20s. Early detection of cancer and appropriate treatment can help extend the lifespan of theseindividuals.
Page 37 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Canavan DiseaseGene: ASPA
Variant Accuracy PrecisionE285A 1.000 1.000Y231X 1.000 1.000A305E 1.000 1.000IVS2-2A>G 1.000 1.000
Detection Rate Population53.37% African American98.00% Ashkenazi Jewish53.37% Eastern Asia53.37% Finland53.37% French Canadian or Cajun53.37% Hispanic53.37% Middle East53.37% Native American53.37% Northwestern Europe53.37% Oceania53.37% South Asia53.37% Southeast Asia53.37% Southern Europe
What is Canavan disease?Canavan disease is an inherited disorder that destroys the myelin sheath, the white matter that insulatesnerve cells in the brain. It causes overall muscle weakness and developmental delay leading to severemental disability. Symptoms usually begin at 3 to 5 months of age with poor muscle tone (hypotonia),which causes problems turning over, controlling head movements, and sitting up. The infant's headalso becomes rapidly larger. Over time, people with the condition become unable to swallow anddevelop sleep disturbances, seizures, and blindness. Most people with Canavan disease die inchildhood, although some have lived into their teens and early twenties.
Canavan disease is caused by a deficiency in an enzyme called aspartoacylase. This enzyme breaksdown a material called N-acetyl-L-aspartic acid (NAA) in the brain. Without enough enzyme, theNAA builds up in the brain and destroys its white matter.
How Common is Canavan disease?The prevalence of Canavan disease in the general population is unknown. Among people of AshkenaziJewish descent, the disease affects approximately 1 in 6,400 to 13,500 people, making 1 in every 40 to58 Ashkenazi Jews a carrier.
Page 38 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is Canavan disease Treated?At this time, there is no cure for Canavan disease. Treatment focuses on keeping the affected personcomfortable with proper nutrition and hydration and controlling seizures with medication.
What is the Prognosis for Someone With Canavandisease?Most people with Canavan disease die in childhood, although some survive into their teens or earlytwenties. In childhood they become severely mentally disabled and lose muscle control.
Page 39 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Carnitine Palmitoyltransferase IADeficiencyGene: CPT1A
Variant Accuracy PrecisionP479L 1.000 1.000G710E 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is carnitine palmitoyltransferase IA deficiency?Carnitine palmitoyltransferase IA deficiency (CPT1A deficiency) is an inherited disease in which thebody cannot process long-chain fatty acids (a type of fat) and turn them into energy. Symptoms occurin severe episodes, often during long periods without eating and/or during times of fever orgastrointestinal illness.
A key symptom of the disease is low blood sugar (hypoglycemia) combined with low blood levels ofketones, a by-product of fat breakdown which is used for energy. Together, these symptoms are knownas hypoketotic hypoglycemia. Prolonged periods of hypoketotic hypoglycemia can lead to loss ofconsciousness or seizures.
Other symptoms of CPT1A deficiency include an enlarged liver, muscle weakness, and damage to theliver, heart, and brain due to excess fatty-acid buildup. If untreated, the disease can be life-threatening.
Pregnant women whose fetus has CPT1A deficiency (and therefore is herself a carrier of CPT1Adeficiency) are at risk of developing a complication called fatty liver of pregnancy. This can causenausea, abdominal pain, fatigue, and frequent thirst and urination. It is potentially life-threatening andrequires aggressive treatment.
Page 40 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Symptoms of CPT1A deficiency usually begin in infancy, but in some cases they appear later in life.
How Common is carnitine palmitoyltransferase IAdeficiency?CPT1A deficiency is extremely rare. Fewer than 50 cases have been identified worldwide. The diseaseis thought to be more common among Hutterite people in the northern United States and Canada aswell as the Inuit people of northern Canada, Alaska, and Greenland.
How is carnitine palmitoyltransferase IA deficiencyTreated?A key goal of treatment is to combat low blood sugar (hypoglycemia). A physician will recommend amodified diet, typically with high-carbohydrate, low-fat foods. Infants will need to eat frequentlyduring the day. A corn starch solution consumed regularly overnight will provide a slow release ofenergy that prevents blood sugar from dipping to dangerously low levels. People with CPT1Adeficiency should never go long periods without eating.
When hypoglycemia does occur, it needs to be quickly treated with an intravenous sugar solution inorder to prevent damage to the brain.
Women who are carriers of CPT1A deficiency and become pregnant should undergo testing for liverenzyme levels, especially during times of fasting or illness.
What is the Prognosis for Someone With carnitinepalmitoyltransferase IA deficiency?After fasting or illness, people with CPT1A deficiency are at risk for life-threatening liver failure.These episodes can also cause permanent damage to the brain and liver. However when the disease iscarefully managed, people with CPT1A deficiency can live fairly normal lives.
Page 41 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Carnitine Palmitoyltransferase II DeficiencyIncluding Infantile Form, Myopathic Form, and Lethal Neonatal Form
Gene: CPT2
Variant Accuracy PrecisionS38fs 1.000 1.000Leu178_Ile186delinsPhe 1.000 1.000Q413fs 1.000 1.000P50H 1.000 1.000S113L 1.000 1.000R124X 1.000 1.000P227L 1.000 1.000R503C 1.000 1.000G549D 1.000 1.000Q550R 1.000 1.000P604S 1.000 1.000Y628S 1.000 1.000R631C 1.000 1.000
Detection Rate Population94.19% African American94.19% Ashkenazi Jewish94.19% Eastern Asia94.19% Finland94.19% French Canadian or Cajun94.19% Hispanic94.19% Middle East94.19% Native American94.19% Northwestern Europe94.19% Oceania94.19% South Asia94.19% Southeast Asia94.19% Southern Europe
What is carnitine palmitoyltransferase II deficiency?Carnitine palmitoyltransferase II deficiency (CPT II deficiency) is an inherited disease in which thebody cannot process long-chain fatty acids and turn them into energy. This is due to a defectiveenzyme.
There are three versions of the disease, listed below, each with a very different profile.
Page 42 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Lethal Neonatal Form
The lethal neonatal form of CPT II deficiency is the most severe form of the disease. Symptoms beginwithin days of birth and include liver failure, respiratory failure, a weakened and/or inflamed heart,irregular heartbeat leading to heart attack, kidney disease, and brain abnormalities. Usually theseinfants die within the first year of life.
Severe Infantile Hepatocardiomuscular Form
Symptoms of the severe infantile hepatocardiomuscular form of CPT II deficiency usually beginbetween 6 months and 2 years of age. They include liver failure, a weakened and/or inflamed heart,seizures, low blood sugar, abdominal pain, headache, muscle weakness in the arms and legs, andirregular heartbeat which can result in sudden death during infancy. Severe episodes are often triggeredby fasting, infection, or fever.
Myopathic Form
The myopathic form of CPT II deficiency is the most common and least severe form of the disease.Symptoms can begin at any time from childhood to one’s 60s. People with the myopathic form of CPTII deficiency experience periodic attacks involving their muscles. These episodes are characterized bymuscle pain and weakness. In many people with the disease, muscle tissue breaks down during theseperiods, causing brown or red-colored urine. Rarely, this can cause kidney problems. These attacks canbe brought on by exercise, exposure to cold, stress, general anesthesia, sleep deprivation, or longperiods of time without eating. Between attacks, people with the myopathic form of CPT II deficiencyare typically normal.
The episodes of muscle pain are usually mild. Some people experience a limited number of severeattacks with long periods of normalcy. A smaller number of people experience frequent muscle painthat impairs their normal activity.
Most commonly, carriers of CPT II deficiency do not have any symptoms of the disease, however asmall number of symptomatic carriers have been reported.
The myopathic form of CPT II deficiency is more often seen in men than women. Studies have shownthe ratio of symptomatic men to women to be as high as 5-to-1. The reason for this gender differentialis not well understood.
How Common is carnitine palmitoyltransferase IIdeficiency?CPT II deficiency is rare. The lethal neonatal form has been documented in 13 families, while thesevere infantile hepatocardiomuscular form has been studied in 20 families. There are more than 200
Page 43 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

known cases of the myopathic form, however scientists believe this form often goes unrecognized,particularly in its mildest cases, and may be more common than studies have indicated.
How is carnitine palmitoyltransferase II deficiencyTreated?There is no cure for CPT II deficiency, and very little can be done to help infants and children with themore severe forms of the disease, other than to treat symptoms as they arise.
For people with the myopathic form, there are recommendations that can help prevent attacks.Frequent meals with a high-carbohydrate, low-fat diet can be helpful. Supplements of carnitine mayalso be recommended. During infection, a physician may recommend infusions of glucose.Circumstances to avoid include strenuous exercise, long periods of time without eating, and extremecold.
During attacks, a person with the myopathic form of CPT II deficiency should drink plenty of fluids toavoid kidney problems.
People with the myopathic form of CPT II deficiency should avoid taking ibuprofen, valproic acid, anddiazepam in high doses. They should also notify their physician before undergoing general anesthesia,as this can provoke an episode of muscle pain and weakness.
What is the Prognosis for Someone With carnitinepalmitoyltransferase II deficiency?Infants with the lethal neonatal form of CPT II deficiency typically die within the first year of life.Infants and children with the severe infantile hepatocardiomuscular form are susceptible to life-threatening heart problems and typically have shortened lifespans with numerous medical problems.
People with the myopathic form of the disease typically live normal lifespans with periodic muscleproblems. This form of the disease is usually manageable and allows for a good quality of life.
Page 44 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Cartilage-Hair HypoplasiaGene: RMRP
Variant Accuracy Precision262G>T 1.000 1.000g.70A>G 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
97.10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is cartilage-hair hypoplasia?Cartilage-hair hypoplasia (CHH) is an inherited condition that causes an affected person to have anextremely small stature with short arms and legs. This is known as short-limbed dwarfism. People withCHH also tend to have fine, sparse hair and abnormal cartilage. Some people with CHH have animpaired immune system, leaving them more susceptible to infection, notably to a severe course ofchicken pox. Some also have low levels of certain white blood cells. Anemia, a lowered number of redblood cells leading to fatigue and weakness, is common in children with CHH, though it usuallydisappears by adulthood. Some people with CHH are at a higher risk for certain cancers including non-Hodgkin’s lymphoma and skin cancer. Symptoms and their severity vary widely among people withthe disease.
How Common is cartilage-hair hypoplasia?Cartilage-hair hypoplasia is rare. It is most common among the Amish population. One study indicatedthat 1 in 19 Amish were carriers of the disease and 1 in 1340 Amish babies were born with the disease.It is also more common in the Finnish population where 1 in 76 is a carrier and 1 in 23,000 babies hasthe disease.
Page 45 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is cartilage-hair hypoplasia Treated?There is no treatment for cartilage-hair hypoplasia. The drug Acyclovir can be useful to treat chickenpox. Infections, particularly those in childhood, should be given close medical attention. Growthhormones may be a possibility for some patients. Those with extreme immunodeficiency may want toconsider bone marrow transplantation to ameliorate this symptom.
What is the Prognosis for Someone With cartilage-hairhypoplasia?People with cartilage-hair hypoplasia can live a normal lifespan. Those with severe immunodeficiencyneed to monitor their health more closely. Opportunistic infections can be fatal, particularly inchildhood.
Page 46 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

ChoroideremiaGene: CHM
Variant Accuracy PrecisionIVS13+2dupT 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
75.00% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is choroideremia?Choroideremia is an inherited condition that causes progressive vision loss. It is far more common inmen than in women. The condition causes tissues in the back of the eye, namely the retina,photoreceptors, and choroid (a network of blood vessels that lies between the retina and the white ofthe eye) to degenerate over time. Night blindness is typically the first symptom, followed by a loss ofperipheral vision. These symptoms typically develop before the age of 20, although the rate ofdegeneration varies greatly from person to person, even among members of the same family.
Eventually the condition affects a person's central vision—essentially what one sees when one looksstraight ahead—though this typically doesn't begin until one's 40s or 50s. Some people withchoroideremia retain functional central vision into their 70s.
Choroideremia does not affect any other system of the body.
How Common is choroideremia?An estimated 1 in 58,000 people have choroideremia. The overwhelming majority of people affectedby choroideremia are male.
Page 47 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is choroideremia Treated?Treatments for choroideremia largely involve improving the person's nutrition and helping the personcope with visual impairments. Fresh fruits and vegetables, an antioxidant supplement, and omega-3fatty acids—provided either through supplements or foods such as fish—are often recommended by aphysician.
Treatments for vision loss are similar to those recommended for any visually-impaired person. Anoptometrist can help the person make the most of the remaining vision. Counseling may be helpful tocope with the emotional effects of living with decreased vision.
What is the Prognosis for Someone With choroideremia?Choroideremia does not affect a person's lifespan. People can live long, productive lives withchoroideremia, albeit with progressive visual impairment.
Page 48 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

CLN5-Related Neuronal CeroidLipofuscinosisGene: CLN5
Variant Accuracy Precision2467AT 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
94.00% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is CLN5-related neuronal ceroid lipofuscinosis?CLN5-related neuronal ceroid lipofusciosis (NCL) is an inherited disease that causes degeneration ofthe brain leading to a progressive loss of mental and motor skills. It causes blindness and leads to anearly death.
Mutations in the CLN5 gene cause a form of NCL that is often referred to as the Finnish variant oflate-infantile neuronal ceroid lipofuscinosis (fLINCL).
Symptoms usually begin between the ages of 4 and 7. By the age of 10, children typically have losttheir vision and develop seizures, mental disability, muscle twitching, and an inability to controlmuscle movements (ataxia). Between the ages of 8 and 11, they lose the ability to walk independently.They will gradually lose their ability to speak and move and will become profoundly mentallydisabled.
Page 49 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is CLN5-related neuronal ceroidlipofuscinosis?CLN5-related NCL is most common in an area of western Finland known as Southern Ostrobothnia.There, 1 in 24 to 44 is a carrier of the disease. In other parts of Finland, studies have found that 1 in385 are carriers in Eastern Finland and 1 in 1000 in the capital of Helsinki. To date, 29 cases ofCLN5-related NCL have been diagnosed in Finland, one in Sweden, and one in the Netherlands.
How is CLN5-related neuronal ceroid lipofuscinosisTreated?There is no treatment for the underlying cause of CLN5-related NCL. Treatments, such as anti-seizuremedication, can only address the symptoms as they arise.
What is the Prognosis for Someone With CLN5-relatedneuronal ceroid lipofuscinosis?The prognosis for people with the disease is poor. They will be profoundly mentally disabled andunable to speak or move some time after the age of 10. The average life expectancy is about 20 years,though the lifespan of people with the disease has ranged from 14 to 39 years.
Page 50 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Congenital Disorder of Glycosylation TypeIaGene: PMM2
Variant Accuracy PrecisionF119L 1.000 1.000R141H 1.000 1.000
Detection Rate Population53.50% African American38.70% Ashkenazi Jewish53.50% Eastern Asia38.70% Finland38.70% French Canadian or Cajun53.50% Hispanic53.50% Middle East53.50% Native American38.70% Northwestern Europe53.50% Oceania53.50% South Asia53.50% Southeast Asia38.70% Southern Europe
What is congenital disorder of glycosylation type Ia?Congenital disorder of glycosylation type Ia (CDG-Ia) is an inherited metabolic disorder that impairsthe production of glycoproteins, which are proteins that have attached carbohydrates. In CDG-Ia, thereis a defect in an enzyme called phosphomannomutase.
CDG-Ia affects many systems of the body, notably the nervous system. The disease causesdevelopmental delay that can lead to mental disability. It can also cause seizures and stroke-likeepisodes. It impairs both the ability to move physically and the ability coordinate that movement.Approximately 20% of infants with the disease die within the first year of life. Most people with CDG-Ia will be wheelchair bound.
A person with CDG-Ia may show some or all of the following symptoms: a failure to grow at thenormal rate; a particular type of limited vision which leads to blindness (retinitis pigmentosa); anunderactive thyroid (hypothyroidism); an enlarged liver and/or liver disease; heart and kidneyproblems; low blood sugar; a decreased ability for blood to clot following an injury; thickened,swollen, pitted skin (peau d'orange); and bone abnormalities. Some people have many of the abovesymptoms while others have few.
Page 51 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

In people with CDG-Ia, certain distinct features are apparent at birth. These may include invertednipples, poor muscle tone, almond shaped eyes which are crossed, a large forehead, an unusualdistribution of body fat, and abnormal genitals. Part of the brain called the cerebellum is often partiallywasted away. Females with the disease often do not reach sexual development.
How Common is congenital disorder of glycosylationtype Ia?CDG-Ia accounts for 70% of the congenital disorders of glycosylation, which combined affect 1 inevery 50,000 to 100,000 births. Cases of CDG-Ia have been reported worldwide, with about halfcoming from Scandinavian countries.
How is congenital disorder of glycosylation type IaTreated?There is no cure for CDG-Ia, however some measures may be taken to improve the lives of peopleaffected by the disease. Parents of a young child with CDG-Ia should ensure the child gets the bestpossible nutrition to help with growth. Some children will require a feeding tube. Early use ofoccupational, physical, and speech therapy may be helpful in improving the child's long-term abilitiesin these areas. Surgical or non-surgical measures may correct crossed eyes and ensure better vision.Infusion of blood plasma may be necessary before a surgery to help clotting. Medications may help tocontrol seizures. Various hormones may be useful if the person has an underactive thyroid gland.Wheelchairs and other movement aids are often useful as well.
What is the Prognosis for Someone With congenitaldisorder of glycosylation type Ia?Twenty percent of people with CDG-Ia die within the first year of life, often due to infection, liverproblems, or heart disease. Others with CDG-Ia may live into adulthood. Most are wheelchair boundthroughout their life. Some are able to speak and converse, albeit with some impairment. People withCDG-Ia are unable to live independently, but may accomplish certain tasks independently.
Page 52 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Congenital Disorder of Glycosylation TypeIbGene: MPI
Variant Accuracy PrecisionR295H 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland>99% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is congenital disorder of glycosylation type Ib?Congenital disorder of glycosylation type Ib (CDG-Ib) is an inherited metabolic disorder that impairsthe production of glycoproteins, which are proteins that have attached carbohydrates. In type Ib, thereis a defect in an enzyme called phosphomannose isomerase.
If left untreated, the disease can cause a wide array of problems including chronic diarrhea, a failure togrow at the expected rate, a loss of protein from the body, vomiting, low blood sugar, difficulty informing blood clots, and liver disease.
CDG-Ib can be effectively treated by taking supplements of mannose, a sugar. With this supplement,life can be relatively normal. Without it, symptoms of the disease can be life-threatening. For thisreason, early diagnosis and treatment is important.
CDG-Ib is distinct from other forms of CDG in that it does not affect the central nervous system.People with CDG-Ib are intellectually normal.
Page 53 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is congenital disorder of glycosylationtype Ib?CDG-Ib is extremely rare, although the exact frequency is unknown.
How is congenital disorder of glycosylation type IbTreated?CDG-Ib is treated with oral supplements of mannose, a sugar. People with CDG-Ib who beginmannose treatment show improvement in most of the symptoms of the disease. Treatment withmannose must be lifelong.
What is the Prognosis for Someone With congenitaldisorder of glycosylation type Ib?With early and regular treatment, a person with CDG-Ib can live a near-normal life. Without it, thedisease can be fatal. Generally, the prognosis will vary depending on the severity of symptoms andtheir response to mannose treatment.
Page 54 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Congenital Finnish NephrosisGene: NPHS1
Variant Accuracy Precision121_122del 1.000 1.000R1109X 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
96.80% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is congenital Finnish nephrosis?Congenital Finnish nephrosis is an inherited disease in which the kidneys are unable to properly filterprotein from the urine due to an abnormality in a protein called nephrin. The disease occurs mainly inpeople of Finnish origin. It is often fatal by the age of five and many cases are fatal within the firstyear. If the child survives to the age of two or three, kidney transplantation may allow for a morenormal lifespan.
Children with congenital Finnish nephrosis are often born prematurely with a low birth weight. Highlevels of protein in the blood, combined with kidney failure, cause the whole body to swell with excessfluids. These children have a poor appetite and urinate less frequently than chidren without the disease.Children with congenital Finnish nephrosis have difficulty getting needed nutrients and may not growas large as they would otherwise.
People with congenital Finnish nephrosis cannot retain sufficient amounts of antibodies that help thebody fight infection. As a result, they are more prone to infection. They are also prone to inappropriateand potentially harmful blood clots.
Symptoms of the disease begin in the first days or weeks after birth, but always before the age of threemonths.
Page 55 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is congenital Finnish nephrosis?As indicated by its name, congenital Finnish nephrosis is fairly common in Finland, where it affects 1in 8,000 births. In the United States, it is rare, but more common in people of Finnish ancestry. Thedisease is extremely common among Old Order Mennonites in Lancaster County, Pennsylvania. It isestimated that 1 in 500 children born in this population are affected by the disease.
How is congenital Finnish nephrosis Treated?Because congenital Finnish nephrosis is often fatal in infancy, early and vigilant treatment is necessaryto allow the child to live until the age of two or three, at which time he or she may receive a kidneytransplant. This is the only hope for a normal lifespan. The disease does not affect the new kidney.
If the disease is too severe, the child's kidneys may need to be removed before he or she is old enoughfor a transplant. Dialysis machines can be used as a stopgap measure to filter wastes from the child'sblood until a transplant can be completed.
A physician may recommend infusions of protein for these children to help replace what is lost in theurine. Diuretic drugs may be prescribed to help eliminate excess water and thus eliminate someswelling. Antibiotics will be necessary to control infection.
Some children with the disease have abnormal thyroid activity and may require hormone replacement.Others have a tendency towards blood clots and may benefit from a blood thinner.
Good nutrition is key to growth. Those who cannot eat sufficient quantities may need a feeding tube.
What is the Prognosis for Someone With congenitalFinnish nephrosis?Many cases of congenital Finnish nephrosis are fatal within five years. If the child lives until the age oftwo or three, an early kidney transplant may help him or her to live a more normal lifespan.
Page 56 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Cystic FibrosisGene: CFTR
Variant Accuracy PrecisionG85E 1.000 1.000R117H 1.000 1.000R334W 1.000 1.000R347P 1.000 1.000A455E 1.000 1.000G542X 1.000 1.000G551D 1.000 1.000R553X 1.000 1.000R560T 1.000 1.000R1162X 1.000 1.000W1282X 1.000 1.000N1303K 1.000 1.000F508del 1.000 1.000I507del 1.000 1.0002184delA 1.000 1.0003659delC 1.000 1.000621+1G>T 1.000 1.000711+1G>T 1.000 1.0001717-1G>A 1.000 1.0001898+1G>A 1.000 1.0002789+5G>A 1.000 1.0003120+1G>A 1.000 1.0003849+10kbC>T 1.000 1.000E60X 1.000 1.000R75X 1.000 1.000G91R 1.000 1.000E92X 1.000 1.000R117C 1.000 1.000Y122X 1.000 1.000G178R 1.000 1.000L206W 1.000 1.000G330X 1.000 1.000T338I 1.000 1.000R347H 1.000 1.000R352Q 1.000 1.000S364P 1.000 1.000G480C 1.000 1.000Q493X 1.000 1.000V520F 1.000 1.000C524X 1.000 1.000
Page 57 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Variant Accuracy PrecisionS549I 1.000 1.000S549N 1.000 1.000Q552X 1.000 1.000A559T 1.000 1.000P574H 1.000 1.000G622D 1.000 1.000R709X 1.000 1.000K710X 1.000 1.000Q890X 1.000 1.000R1066C 1.000 1.000R1070Q 1.000 1.000W1089X 1.000 1.000Y1092X 1.000 1.000M1101K 1.000 1.000D1152H 1.000 1.000R1158X 1.000 1.000S1196X 1.000 1.000S1235R 1.000 1.000Q1238X 1.000 1.000S1251N 1.000 1.000S1255X 1.000 1.000R1283M 1.000 1.000dele2-3 21kb 1.000 1.0003199del6 1.000 1.000F311del 1.000 1.000394delTT 1.000 1.000574delA 1.000 1.000663delT 1.000 1.000935delA 1.000 1.000936delTA 1.000 1.0001078delT 1.000 1.0001161delC 1.000 1.0001609delCA 1.000 1.0001677delTA 1.000 1.0001949del84 1.000 1.0002043delG 1.000 1.0002055del9>A 1.000 1.0002105-2117del13insAGAAA 1.000 1.0003171delC 1.000 1.0003667del4 1.000 1.0003821delT 1.000 1.0003876delA 1.000 1.0001288insTA 1.000 1.0002184insA 1.000 1.000
Page 58 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Variant Accuracy Precision2307insA 1.000 1.0002869insG 1.000 1.0003905insT 1.000 1.000296+12T>C 1.000 1.000405+1G>A 1.000 1.000405+3A>C 1.000 1.000406-1G>A 1.000 1.000711+5G>A 1.000 1.000712-1G>T 1.000 1.0001811+1.6kbA>G 1.000 1.0001812-1G>A 1.000 1.0001898+1G>T 1.000 1.0001898+5G>T 1.000 1.0003272-26A>G 1.000 1.0003120G>A 1.000 1.000457TAT>G 1.000 1.0002183AA>G 1.000 1.000IVS8-5T 1.000 1.000I148T 1.000 1.000I506V 1.000 1.000F508C 1.000 1.000S549R(T>G) 1.000 1.000W1204X(c.3611G>A) 1.000 1.000S549R(A>C) 1.000 1.000W1204X(c.3612G>A) 1.000 1.000
Detection Rate Population80.00% African American97.00% Ashkenazi Jewish56.11% Eastern Asia90.79% Finland91.29% French Canadian or Cajun83.77% Hispanic36.49% Middle East
<10% Native American89.69% Northwestern Europe
<10% Oceania49.49% South Asia56.11% Southeast Asia89.69% Southern Europe
Page 59 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
What is cystic fibrosis?Cystic fibrosis (CF) is a genetic condition characterized by the production of abnormally thick, stickymucus, particularly in the lungs and digestive system. While it is normal to have mucus lining theorgans of the respiratory, digestive, and reproductive systems in order to lubricate and protect them, inpeople with CF this mucus is thick and sticky. This abnormal mucus results in the clogging andobstructing of various systems in the body. CF is a chronic condition that worsens over time.
Most people with CF experience breathing problems and frequent lung infections that lead topermanent lung damage such as scarring (fibrosis) and sac-like growths (cysts). The pancreas, an organthat produces insulin and digestive enzymes, is often affected by CF. The sticky mucus caused by CFcan block ducts which ferry enzymes from the pancreas to the rest of the body, resulting in problemssuch as diarrhea, malnutrition, and poor growth. Infertility, particularly in men, and delayed pubertyare also common among people with cystic fibrosis.
The severity of symptoms varies from person to person. Most cases of CF are diagnosed in earlychildhood. Those who are diagnosed after the age of 18 typically have a milder form of the condition.
Mutations in the same gene that causes CF can result in a condition in males called congenital absenceof the vas deferens (CAVD). In CAVD, the vas deferens (a reproductive organ involved in spermtransport) is improperly formed, leading to infertility.
How Common is cystic fibrosis?According to the National Institutes of Health, CF is the most common deadly inherited conditionamong Caucasians in the United States. Disease-causing mutations in the CFTR gene are morecommon in some ethnic populations than others.
Ethnic Group Carrier Rate Affected Rate
Caucasian 1 in 28 1 in 3,000
Ashkenazi Jewish 1 in 28 1 in 3,000
Hispanic 1 in 46 1 in 8,300
African American 1 in 66 1 in 17,000
Asian 1 in 87 1 in 30,000
Page 60 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is cystic fibrosis Treated?There is no treatment that addresses the cause of CF, but there are many options to treat the symptomsit produces. Because thick mucus can build up in the respiratory system, it is important to keep theperson's airways open in order to ease breathing and prevent infection. This can be accomplished withvarious prescription drugs as well as by physically loosening mucus by pounding on the person's backin a prescribed way. This treatment, known as "postural drainage and chest percussion" must beperformed by someone other than the affected person, and is typically done at least once daily. Asrespiratory infections occur, physicians typically prescribe antibiotics.
Physicians will also monitor the digestive system to ensure that the person is getting proper nutrition.Enzymes or vitamin supplements may be prescribed. Both the respiratory and digestive systems of aperson with CF must be monitored regularly by his or her medical team.
Surgery may be needed to correct certain problems caused by CF. Lung transplants are an option forsome people.
What is the Prognosis for Someone With cystic fibrosis?Thanks to improved treatments and a better understanding of the condition, the average life expectancyfor people with CF who live to adulthood is 35 years. Children born with CF today who receive earlytreatment may live even longer.
Page 61 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

CystinosisGene: CTNS
Variant Accuracy Precision537del21 1.000 1.000W138X 1.000 1.000L158P 1.000 1.000D205N 1.000 1.000
Detection Rate Population<10% African American
16.62% Ashkenazi Jewish<10% Eastern Asia
16.62% Finland60.91% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
16.62% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
16.62% Southern Europe
What is cystinosis?Cystinosis is an inherited disease that causes the amino acid cysteine to accumulate within body cellsand form crystals which can damage the body’s organs, particularly the kidneys and eyes. Withouttreatment, children with the condition will experience kidney failure around the age of 10.
There are three forms of cystinosis. The most severe form, nephropathic cystinosis, appears in infants.It causes poor growth and renal tubular Fanconi syndrome, a kidney disorder in which the organeliminates certain essential nutrients and minerals. The loss of these nutrients inhibits normal bodygrowth and may result in soft, bowed bones. Cysteine crystals also accumulate in the eyes, causingphotophobia, an extreme sensitivity to light. Other symptoms may include muscle wasting, difficultyswallowing, diabetes, an underactive thyroid gland, and nervous system problems.
Less severe forms of the disease cause symptoms to begin later in life and may not affect the kidneys.
How Common is cystinosis?Cystinosis affects approximately 1 in 200,000 people. The disease is most common in Brittany, France,where it affects 1 in 26,000.
Page 62 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is cystinosis Treated?Thanks to a drug called cysteamine, cystinosis has become easier to manage. Taken orally in capsules(brand name: Cystagon), it reduces the accumulation of cysteine crystals in the body. The drug hasbeen shown to delay or prevent kidney failure and improve growth rates in children. Cysteamine eyedrops have been successful in relieving photophobia in people with cystinosis, although they are notyet approved by the FDA for that purpose.
Nutritional monitoring is important in children with cystinosis. These children require a large amountof water to prevent dehydration. Supplements of several vitamins and minerals are also recommendedfor most people with the disease. Human growth hormone treatments have been shown to help peoplewith cystinosis reach normal height.
Kidney transplants are an option for people with cystinosis. Cysteine crystals will not build up in thenewly transplanted kidney, although they may still affect other organs of the body.
What is the Prognosis for Someone With cystinosis?Cystagon has extended the lifespan of people with cystinosis, but exact lifespan is not known. Somepeople with the disease have lived into their 50s.
Page 63 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Factor V Leiden ThrombophiliaGene: F5
Variant Accuracy PrecisionR506Q 1.000 1.000H1299R 1.000 1.000D2222G 1.000 1.000
Detection Rate Population>99% African American>99% Ashkenazi Jewish>99% Eastern Asia>99% Finland>99% French Canadian or Cajun>99% Hispanic>99% Middle East>99% Native American>99% Northwestern Europe>99% Oceania>99% South Asia>99% Southeast Asia>99% Southern Europe
What is factor V Leiden thrombophilia?Factor V Leiden thrombophilia is an inherited disease which increases the chances of developingpotentially dangerous blood clots. These blood clots tend to develop in the veins of the leg (deep veinthrombosis), but other types of blood clots are also possible.
Please note that most people who have the disease do not get abnormal blood clots. In the generalpopulation, 1 in 1,000 people develop a blood clot each year. In people with one mutation for factor VLeiden thrombophilia (and one normal copy of the gene, making them a carrier), that risk increases tobetween 4 and 8 in 1,000. This is comparable to the 3 to 6 fold increased risk of blood clots whentaking an estrogen-containing birth control pill, which every obstetrician is already required to informwomen about prior to taking the pill. In people with two mutations causing factor V Leidenthrombophilia, the risk increases substantially to between 18 and 80 in 1,000.
Certain factors increase the risk of blood clots in people with factor V Leiden thrombophilia. Thesecan include smoking, advanced age, obesity, oral contraceptives, hormone replacement therapy, airtravel, pregnancy, organ transplantation, surgery, cancer, and the presence of other genetic bloodclotting disorders. Children with factor V Leiden thrombophilia rarely develop abnormal blood clots.Typically if these abnormal clots occur, they first appear in adulthood.
Page 64 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Pregnant women who have two genetic mutations that cause factor V Leiden thrombophilia are at anincreased risk for certain complications including miscarriage, high blood pressure (preeclampsia),delayed physical development of the fetus, and a separation of the placenta from the uterine wall. Theirrisk of losing a pregnancy is 2 to 3 times greater than the general population. Please note however thatmost women with the disease will have normal pregnancies.
How Common is factor V Leiden thrombophilia?Roughly 1 in 5,000 people in the U.S. and Europe have two copies of the mutation that causes factor VLeiden thrombophilia, putting them at higher risk for abnormal blood clots.
Having one copy of the mutation that causes factor V Leiden thrombophilia (and one normal copy ofthe gene) is fairly common in the United States and Europe. Between 3 and 8% of Caucasians haveone copy of the mutation. (Please note that this causes only a slightly elevated risk for abnormal clots.)
How is factor V Leiden thrombophilia Treated?A physician with knowledge of a person's overall health can help guide him or her in finding theappropriate treatment for factor V Leiden thrombophilia. For people with recurrent abnormal clots,long-term use of preventive medication may be recommended. For people with two factor V Leidenthrombophilia mutations who do not have a history of clotting, long-term use of medication may berecommended, although it may lead to a higher risk for excessive bleeding.
People with only one copy of the factor V Leiden thrombophilia mutation (and one normal gene)typically do not use any preventive medications, as the risks for excessive bleeding are seen tooutweigh the anti-clotting benefits. During short periods of higher risk, such as surgery, trauma, orpregnancy, medication may be prescribed.
When clots are discovered, they are often treated with medication according to normal medicalprotocols. Women with deep vein thrombosis may be asked to wear compression stockings for a periodof time following the clot.
People with factor V Leiden thrombophilia may want to avoid smoking, oral contraceptives, hormonereplacement therapy, and obesity.
What is the Prognosis for Someone With factor V Leidenthrombophilia?People with two mutations for factor V Leiden thrombophilia are at an 18 to 80 times greater risk thanthe general population for life-threatening blood clots. That said, the majority of people with thecondition do not experience life-threatening blood clots and will live normal lifespans.
Page 65 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Studies have shown that people with only one mutation for factor V Leiden thrombophilia have anormal lifespan.
Page 66 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Factor XI DeficiencyGene: F11
Variant Accuracy PrecisionIVS14+1G>A 1.000 1.000IVS14del14 1.000 1.000E117X 1.000 1.000F283L 1.000 1.000
Detection Rate Population<10% African American>99% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is factor XI deficiency?Factor XI deficiency, also called factor 11 deficiency or hemophilia C, is an inherited disorder which isusually mild but can cause uncontrolled bleeding. This bleeding tends to be more severe after surgery,injury, or childbirth. Bleeding can be a particular problem after tooth extraction, dental surgery, tonsilsurgery, or urinary tract surgery.
People with factor XI deficiency may also be prone to bruising, nosebleeds, or blood in their urine.Rarely, male children with the disease will bleed heavily following circumcision. More than half ofwomen with the disease have abnormally heavy and prolonged menstrual periods.
It is uncommon for people with factor XI deficiency to bleed spontaneously for no obvious reason.There may be a delay in bleeding following an injury.
Factor XI is a protein produced by the liver and found in the blood. It helps platelets in the blood toclot following an injury to a blood vessel. In people with factor XI deficiency, levels of factor XI arelower than normal. Bleeding problems tend to occur when factor XI levels are lower than 15% of thenormal level. Bleeding problems can occur, however, when levels are as high as 70% of the normal
Page 67 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

level. The severity of the bleeding varies widely from person to person, even among members of thesame family.
Carriers of factor XI deficiency are at elevated risk for bleeding problems. Studies have suggested that20 to 50% of carriers of the disease show "excessive bleeding," although the definition of this phrasevaries. Rarely, carriers have shown major bleeding problems.
How Common is factor XI deficiency?Factor XI deficiency is fairly common among Ashkenazi Jews. One in eight Ashkenazi Jews is thoughtto be a carrier of factor XI deficiency. In one study, 1 in 190 Ashkenazi Jews has a severe bleedingproblem related to factor XI deficiency, while a different study found a much lower rate of severesymptoms, 1 in 450. Among non-Jews in the United States, only 1 in 1,000,000 have factor XIdeficiency, making it very rare.
The disease is also common among families in northwest England, where 1 in 10,000 people has thedisease. Other groups at greater risk for carrying mutations that cause factor XI deficiency are IraqiJews, Sephardic Jews, and people of Arab background living in Israel.
How is factor XI deficiency Treated?In the United States, factor XI deficiency is treated with infusions of fresh frozen blood plasma. Thisblood plasma contains normal quantities of factor XI, thus temporarily enhancing the body’s ability toclot. However, significant amounts of plasma may be required to achieve the desired clotting effectdue to the low concentration of factor XI in plasma.
In Europe, there are several commercially available concentrated doses of factor XI. One ismanufactured in the United Kingdom, the other in France.
In the case of bleeding in the mouth, nose, intestines, or uterus, there are several medications whichmay be helpful, though they are not effective for major internal bleeding and can cause clottingthroughout the body.
What is the Prognosis for Someone With factor XIdeficiency?Factor XI deficiency is not known to affect lifespan. In people who do not realize they have thedisease, life-threatening bleeding is possible following surgery or injury.
Page 68 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Familial DysautonomiaGene: IKBKAP
Variant Accuracy PrecisionIVS20+6T>C 1.000 1.000P914L 1.000 1.000R696P 1.000 1.000
Detection Rate Population<10% African American>99% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is familial dysautonomia?Familial dysautonomia is an inherited condition that causes nerve cells to deteriorate. It affects theautonomic nervous system, which controls involuntary actions such as breathing, tear production,blood pressure, and body temperature. It also affects the sensory nervous system, which controlssenses such as the abilities to perceive taste, pressure, pain, and temperature.
Early symptoms in infants include feeding problems, poor growth, lack of tears, poor muscle tone,frequent lung infections, and marked fluctuations in body temperature. Until about age 6, children withthe condition may also hold their breath for long periods of time, which may cause fainting or maketheir lips or skin appear blue. They may learn to walk and talk later than average.
Starting around age 5 or 6, children with the condition may develop symptoms including bed-wetting,vomiting, reduced sensitivity to temperatures and pain, decreased ability to taste, poor balance,abnormal curvature of the spine, easily fractured bones, and kidney and heart problems. Theycommonly experience a sharp drop in blood pressure when they stand up, which can cause blurredvision, dizziness, or fainting. They may also have episodes of high blood pressure when nervous orexcited.
Page 69 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

By adulthood, people with familial dysautonomia may have balance problems that prevent them fromwalking unaided. Other common complications include sleep apnea, lung damage due to repeatedinfections, poor vision as optic nerves atrophy, and kidney disease. Intellect is not usually impaired.
How Common is familial dysautonomia?Familial dysautonomia is found almost exclusively in people of Ashkenazi Jewish descent, where itaffects approximately 1 in 3,700 people. Roughly 1 in 31 Ashkenazi Jews is a carrier of the disease. Itis extremely rare in the general population.
How is familial dysautonomia Treated?There is no cure for the cause of familial dysautonomia. Treatment focuses on relieving its symptoms.
Infants with the condition may need to be fed thickened formula to ensure adequate nutrition andprevent them from inhaling their food. Vomiting crises are treated with IV fluids and anti-nauseamedication. Recurrent pneumonia caused by inhaling food or vomit requires daily chest physiotherapy.Older children who experience low blood pressure may require elastic stockings and leg exercises toimprove muscle tone and prevent blood from pooling in leg veins. Corneal injuries caused by low tearproduction may be treated with regular eye drops, soft contact lenses, or in rare cases, surgery. Spinalfusion surgery may be necessary to correct scoliosis. Kidney disease may require dialysis. Sleep apneais generally treated with a machine to support breathing. Many adults require walkers or wheelchairs.
What is the Prognosis for Someone With familialdysautonomia?The average lifespan of a person with familial dysautonomia is significantly shortened. Only 60% ofpeople with the disease survive to age 20.
Page 70 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Familial Mediterranean FeverGene: MEFV
Variant Accuracy PrecisionI692del 1.000 1.000T267I 1.000 1.000F479L 1.000 1.000R653H 1.000 1.000M680I 1.000 1.000M694I 1.000 1.000K695R 1.000 1.000A744S 1.000 1.000R761H 1.000 1.000P369S 1.000 1.000R408Q 1.000 1.000M694V 1.000 1.000V726A 0.997 1.000
Detection Rate Population68.61% African American69.30% Ashkenazi Jewish68.61% Eastern Asia68.61% Finland68.61% French Canadian or Cajun68.61% Hispanic68.61% Middle East68.61% Native American68.61% Northwestern Europe68.61% Oceania68.61% South Asia68.61% Southeast Asia68.61% Southern Europe
What is familial Mediterranean fever?Familial Mediterranean fever (FMF) is an inherited condition which causes episodic attacks of feverand painful inflammation of the abdomen, chest, and joints. People with FMF may also develop a rashduring these attacks. The attacks last for 1 to 3 days and can vary in severity. Between attacks, theperson typically feels normal. These symptom-free periods can last for days or even years.
In 80-90% of people affected by FMF, the first attack occurs by the age of 20. Less commonly,symptoms begin later in life. Children who have FMF may experience periodic fever as their onlysymptom.
Page 71 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Some people with FMF develop a protein buildup in various parts of the body, notably the kidney. Ifleft untreated, this can lead to life-threatening kidney failure. People who do not experience thecharacteristic attacks of FMF can still develop this particular form of kidney failure. This symptom ismost common among people of Turkish and North African Jewish heritage, affecting 60% and 75%respectively.
Other symptoms that can occur during an attack of FMF include headache and inflammation of theheart and/or testicles. Affected people may also develop an inflammation of the membrane thatsurrounds the brain and spinal cord, though this is not usually serious or damaging. People with FMFwho go untreated may experience decreased fertility.
About half of people with FMF have mild symptoms preceding an attack. These may include a mild,unpleasant sensation in parts of the body that will soon be affected or may consist of other physical andemotional symptoms.
How Common is familial Mediterranean fever?FMF is most common among ethnic groups from the Mediterranean region, notably people ofArmenian, Arab, Turkish, Iraqi Jewish, and North African Jewish ancestry. One in every 200 to 1,000people in these groups is affected by the disease and carrier rates in some populations have beenestimated as high as 1 in 5.
Cases of FMF have also been found in other populations, including Italians, Greeks, Spaniards,Cypriots, and less commonly, Northern Europeans and Japanese.
How is familial Mediterranean fever Treated?There is no cure for FMF, however the drug colchicine has been very effective in preventing thedisease’s characteristic attacks. With daily doses of colchicine, 75% of people with FMF can avoidattacks with an additional 15% showing an improvement in their symptoms. Colchicine also preventsthe dangerous buildup of proteins in the kidneys which could otherwise lead to kidney failure.
Episodic attacks of fever and inflammation can be treated with non-steroidal anti-inflammatory drugs.Those who do develop serious kidney failure may be helped by kidney transplantation.
What is the Prognosis for Someone With familialMediterranean fever?With early and regular treatment, people with FMF can live a normal lifespan and may even besymptom-free. The disease has the potential to be life-threatening only if the person is untreated (ordoes not respond to treatment) and develops kidney failure.
Page 72 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Fanconi Anemia Type CGene: FANCC
Variant Accuracy PrecisionIVS4+4A>T 1.000 1.000322delG 1.000 0.987Q13X 1.000 1.000R548X 1.000 1.000
Detection Rate Population59.80% African American99.00% Ashkenazi Jewish59.80% Eastern Asia59.80% Finland59.80% French Canadian or Cajun59.80% Hispanic59.80% Middle East59.80% Native American59.80% Northwestern Europe59.80% Oceania59.80% South Asia59.80% Southeast Asia59.80% Southern Europe
What is Fanconi anemia type C?Fanconi anemia type C is an inherited disorder in which the body cannot properly produce a proteinthat protects DNA from damage. This defective protein results in an impaired ability of bone marrowto produce all types of blood cells. Without a sufficient number of red blood cells, the body does notreceive enough oxygen, which can lead to abnormal bones and organs as well as developmental delay.A shortage of white blood cells makes the body more susceptible to infection and cancer. A reductionin blood platelets make it difficult for the blood to clot when an injury arises.
In many cases, the first symptoms of Fanconi anemia type C appear in infancy as frequent nosebleeds,a tendency to bruise, and physical abnormalities such as spotted skin or malformations of the thumbs,forearms, eyes, kidneys, gastrointestinal system, ears, or heart. Children with the disease may alsoshow signs of hearing loss or developmental delay. However 25 to 40% of people with the conditiondo not have physical abnormalities. They may be first diagnosed in childhood with abnormally lowlevels of red blood cells, white blood cells, or platelets. Although their bone marrow may appearnormal at first, it deteriorates progressively. Most people with the disease are diagnosed by age 12.
Because Fanconi anemia type C prevents cells from repairing themselves when the DNA is damaged,people with the condition are at higher than average risk of cancer. Occasionally, the initial signs of
Page 73 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

leukemia appear in childhood as the first symptom of the disease. Other cancers may also appear at anunusually early age, particularly tumors of the head and neck, esophagus, cervix, vulva, or liver. Thesecancers commonly develop in the early 20s.
Certain mutations are associated with more or less severe courses of Fanconi anemia type C. Forexample, one particular mutation is associated with less severe symptoms in people of Japaneseancestry.
How Common is Fanconi anemia type C?Fanconi anemia type C affects approximately 1 in 100,000 people. In the general population, 1 in 300people are carriers of mutations that cause this disease. Fanconi anemia is most common in people ofAshkenazi Jewish descent, where 1 in 90 are carriers and 1 in 32,000 have the disease.
How is Fanconi anemia type C Treated?There is currently no cure for Fanconi anemia type C. Treatment consists of watching for symptomsand treating them as they appear.
About half of all people with the condition can improve their blood cell counts with medication. Over aperiod of years, however, people often develop resistance to the medication. Treatment withmedication may also decrease the effectiveness of a later bone marrow transplant.
Bone marrow transplantation can cure the leukemia associated with Fanconi anemia type C. Howeverpeople with the condition are extremely sensitive to the chemotherapy and the radiation treatmentnecessary to prepare for transplantation, so they may not be good candidates for this surgery. A bonemarrow transplant does not prevent solid tumors elsewhere in the body, which must be treated withchemotherapy and radiation.
People with Fanconi anemia type C must undergo regular blood cell counts, bone marrow biopsies,liver scans, and gynecological, dental, and rectal exams to detect early-stage cancers so they can beremoved as soon as possible.
What is the Prognosis for Someone With Fanconi anemiatype C?Most people with Fanconi anemia type C die before the age of 30. A bone marrow transplant canextend lifespan.
Page 74 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Fumarase DeficiencyGene: FH
Variant Accuracy Precision1431_1433dupAAA 1.000 1.000
Detection Rate Population25.00% African American25.00% Ashkenazi Jewish25.00% Eastern Asia25.00% Finland25.00% French Canadian or Cajun25.00% Hispanic25.00% Middle East25.00% Native American25.00% Northwestern Europe25.00% Oceania25.00% South Asia25.00% Southeast Asia25.00% Southern Europe
What is fumarase deficiency?Fumarase deficiency is an inherited disease that primarily affects the brain and nervous system. Peoplewith the disease often have a small head, abnormal brain structure, and severe mental disability. Theyalso typically have poor muscle tone and do not grow and gain weight at the expected rate.
Commonly, although not always, people with fumarase deficiency have characteristic facial featuresincluding a prominent forehead, small jaw, wide-set eyes, and sunken nasal bridge. Many experienceseizures, heartbeat abnormalities, an enlarged liver and spleen, inflammation of the pancreas, and anexcess of and/or deficiency in certain types of blood cells.
The disease is caused by a defect in an enzyme called fumarase which helps cells use oxygen andproduce energy.
How Common is fumarase deficiency?Fumarase deficiency is extremely rare, with roughly 100 cases having been reported worldwide. It ismost common in an isolated community of the Fundamentalist Church of Jesus Christ of Latter-DaySaints living on the Utah-Arizona border.
Page 75 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is fumarase deficiency Treated?There is no cure or effective treatment for fumarase deficiency. A feeding tube may be necessary forchildren unable to get proper nutrition by mouth. Physical therapy and wheelchairs may also behelpful.
What is the Prognosis for Someone With fumarasedeficiency?Most people with fumarase deficiency live only a few months after birth or die in childhood. Somehave lived into early adulthood with severe mental and motor disability.
Page 76 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
GalactosemiaGene: GALT
Variant Accuracy PrecisionIVS2-2A>G 1.000 1.000T138M 1.000 1.000F171S 1.000 1.000Q169K 1.000 1.000L195P 1.000 1.000Y209C 1.000 1.000K285N 1.000 1.000X380R 1.000 1.000S135L 1.000 1.000Q188R 1.000 1.000
Detection Rate Population62.00% African American70.00% Ashkenazi Jewish
<10% Eastern Asia70.00% Finland70.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
70.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
70.00% Southern Europe
What is galactosemia?Galactosemia is a treatable inherited disease that reduces the body's ability to metabolize galactose, asimple sugar found in milk. Classic galactosemia, for which Counsyl screens, can be fatal withoutprompt treatment and careful management. Because milk is a staple of an infant's diet, diagnosis andtreatment within the first week of life is critical to avoiding mental disability and life-threateningcomplications.
Classic galactosemia, the severe form of the disease, is caused by a deficiency in the enzymegalactose-1-phosphate uridyltransferase. People with classic galactosemia have less than 5% of thenormal activity of this enzyme. After only a few days of drinking milk, including breast milk, an infantwith classic galactosemia will show symptoms including loss of appetite, jaundice, vomiting, lethargy,and convulsions. Without immediate and vigilant lifelong treatment, children with the condition will
Page 77 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

experience life-threatening complications such as severe infections, cirrhosis of the liver, and mentaldisability. Even with treatment, children can still develop cataracts, speech problems, stunted growthand motor function, and learning disabilities. Most females will eventually develop menstrualirregularities and go through premature menopause.
Please note that galactosemia is not the same as lactose intolerance, a more common and less seriouscondition.
How Common is galactosemia?Classic galactosemia affects approximately 1 in 30,000 newborns.
How is galactosemia Treated?People with classic galactosemia must monitor their galactose levels with regular blood tests andfollow a lifelong diet free of milk, milk products, or other foods containing galactose and lactose.Infants should be fed with lactose-free formulas such as soy formula or Nutramigen, a hypoallergenicformula with no lactose or soy. As children learn to feed themselves, parents must teach them how toread product labels so they can avoid any food containing milk, dry milk, and milk products. Oftengalactosemic people require calcium supplements to avoid calcium deficiency.
People with galactosemia should work with a nutritionist to determine the best course of treatment.
What is the Prognosis for Someone With galactosemia?Most people who are diagnosed early with classic galactosemia and who carefully follow a galactose-free diet can have a normal lifespan. They are still at risk, however, for cataracts, speech defects, poorgrowth, poor intellectual function, neurologic deficits and ovarian failure. If the treatment of classicgalactosemia is not prompt and consistent, life-threatening complications and irreversible mentaldisability can result.
Page 78 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Gaucher DiseaseGene: GBA
Variant Accuracy PrecisionN370S 1.000 1.000L444P 1.000 1.0001035insG 1.000 1.000IVS2+1G>A 1.000 1.000V394L 1.000 1.000D409V 1.000 1.000R463C 1.000 1.000R463H 1.000 1.000R496H 1.000 1.000
Detection Rate Population70.84% African American95.00% Ashkenazi Jewish68.94% Eastern Asia70.84% Finland70.84% French Canadian or Cajun70.84% Hispanic70.84% Middle East70.84% Native American70.84% Northwestern Europe70.84% Oceania70.84% South Asia68.94% Southeast Asia70.84% Southern Europe
What is Gaucher disease?Gaucher disease is an inherited condition in which the body fails to properly produce a particularenzyme needed to break down a fatty substance called glucocerebroside. Without this enzyme,glucocerebroside and several other associated substances will build up in the body and reach toxiclevels, damaging different organs of the body.
There are five main types of Gaucher disease, each with different symptoms. These types are describedbelow.
Type 1
Type 1 is the most common form of the disease. It can affect people at any age and its symptoms varywidely from mild to severe.
Page 79 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Most people with type 1 Gaucher disease have symptoms related to their bones. These may includebone pain, fractures, and arthritis. On the mild end of the spectrum, some experience only a small dropin their bone mineral density leaving them without symptoms. In more severe cases, the bones can losetheir blood supply and begin to collapse. Bone problems are often the most debilitating aspect of thedisease.
People with type 1 Gaucher disease often have an enlarged liver and spleen. They also have a lowerednumber of red blood cells and platelets. With fewer red blood cells, a person with the disease will oftenbe tired and weak (anemic). Fewer platelets in the blood will make him or her prone to bruising andexcessive bleeding. Lung disease is another common symptom.
Type 1 is distinct from other forms of Gaucher disease in that it usually does not affect the patient'sbrain or spinal cord.
Type 2
Type 2 is known as the infantile or acute neuropathic form of Gaucher disease. Symptoms usuallyappear before age 2 and progress rapidly. People with type 2 Gaucher disease have some of thesymptoms of type 1. These include:
• Enlarged liver and spleen• Lowered number of red blood cells and platelets leading to weakness and tiredness (anemia)• Lung disease
While type 2 is distinct from types 1 and 3 in that it does not cause bone problems, type 2 does causeneurological problems. Neurological symptoms often include limited cognitive and motordevelopment, brainstem abonormalities that can cause breathing problems and difficulty swallowing,constant arching of the back and tilting back of the head, uncontrollable tightening and releasing of themuscles, and the inability to open the mouth. As the nervous system deteriorates, the child maydevelop dementia and the inability to coordinate his or her own movement.
In severe cases, death may occur in utero or shortly before or after birth. In most cases, the child diesbetween the ages of 2 and 4.
Type 3
Type 3 Gaucher disease is known as the juvenile or chronic neuropathic form. Symptoms often beginbefore age 2, though this is variable. Usually the symptoms associated with type 3 progress moreslowly than with type 2. While some people with type 3 Gaucher disease die in childhood, others canlive into their 30s or 40s.
People with type 3 Gaucher disease have the symptoms of type 1. These include:
• Enlarged liver and spleen
Page 80 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

• Lowered number of red blood cells and platelets leading to weakness and tiredness (anemia)• Lung disease• Bone problems including include pain, fractures, and arthritis
As with type 2, type 3 Gaucher disease also causes neurological problems. These may include seizureswhich worsen over time, progressive cognitive problems, and difficulty controlling eye movement.Towards the end of their lives, people with type 3 may also develop dementia.
Perinatal-Lethal Form
The perinatal-lethal form is the most severe form of Gaucher disease. This form usually leads to deathin utero or shortly after birth. Infants with the disease have symptoms including enlarged liver andspleen, lowered number red blood cells and platelets, neurological problems, skin abnormalities, andoften distinct facial features.
Cardiovascular Form
As the name implies, the cardiovascular form of Gaucher disease causes symptoms involving the heart,notably a hardening of the mitral and aortic valves. If this symptom is severe, heart valve replacementmay be required. The cardiovascular form of the disease is sometimes called type 3C.
In addition, the cardiovascular form causes symptoms including:
• A slightly enlarged liver and spleen• Bone problems including include pain, fractures, and arthritis• Difficulty controlling eye movement• A clouding of the eye's cornea, which can affect vision
How Common is Gaucher disease?Type 1 Gaucher disease is the most common form of this disease. It is particularly prevalent amongpeople of Ashkenazi Jewish descent, of whom 1 in 855 is affected by the disease and 1 in 16 is acarrier. In the general population, researchers believe that about 1 in 50,000 will be affected.
Other forms of Gaucher disease are more rare. Type 2 occurs in roughly 1 in 100,000 births. A form oftype 3 is most common among people in the Norrbottnian region of Sweden, where 1 in 50,000 areaffected.
How is Gaucher disease Treated?Enzyme replacement therapy is one of the mainstays of treating Gaucher disease. The drugimiglucerase (brand name: Cerezyme) helps eliminate the buildup of toxic substances which causesome of the disease's symptoms. While it does not improve or prevent any of the neurological
Page 81 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

symptoms found in types 2 and 3, the treatment has been successful in some people to prevent andeven reverse bone and organ damage caused by the disease. While the treatment has only beenavailable since 1991, scientists hope that by preventing bone and organ damage in childhood, peoplewith the Gaucher disease will experience fewer symptoms and live longer lives.
For adults who cannot tolerate Cerezyme, the drug miglustat (brand name: Zavesca) is FDA approvedas an alternative. It can only be used in adults.
People with the cardiovascular form of Gaucher disease often need heart valve replacements, afterwhich Cerezyme can be helpful.
Additional treatments for the symptoms of Gaucher disease include blood transfusions for tirednessand excessive bleeding, joint replacement to relieve pain and restore movement, medication to treatbone pain, and removal of all or part of the spleen.
What is the Prognosis for Someone With Gaucherdisease?Because symptoms of the Gaucher disease vary widely in type and severity, both among the differenttypes and among people with the same type, the outlook is similarly varied.
Those with a milder form of type 1 may have only slight symptoms and can live a normal lifespan.People with severe cases of type 1 Gaucher disease experience debilitating symptoms and may die inchildhood.
Those with type 2 Gaucher disease often die between the ages of 2 and 4, having developed minimalmental and motor skills. In the most severe cases, they may die before or shortly after birth.
People with type 3 Gaucher disease usually develop symptoms in childhood which worsen moreslowly over time. While some have died in childhood, others have lived into their 30s and 40s.
With the perinatal-lethal form, death occurs before or shortly after birth.
For those with the cardiovascular form of the disease, the prognosis depends upon the success of valvereplacement surgery.
Women with milder cases of Gaucher disease have had successful pregnancies.
Page 82 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

GJB2-Related DFNB 1 NonsyndromicHearing Loss and DeafnessGene: GJB2
Variant Accuracy Precision35delG 1.000 1.000167delT 1.000 1.000313del14 1.000 1.000E120del 1.000 1.000W24X 1.000 1.000V37I 1.000 1.000W77R 1.000 1.000W77X 1.000 1.000Q124X 1.000 1.000R184P 1.000 1.000M34T 1.000 1.000
Detection Rate Population<10% African American
90.00% Ashkenazi Jewish<10% Eastern Asia
46.13% Finland46.13% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
46.13% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
46.13% Southern Europe
What is GJB2-related DFNB 1 nonsyndromic hearing lossand deafness?DFNB1 nonsyndromic hearing loss and deafness is an inherited condition in which a person has mildto severe hearing loss from birth. The condition is not progressive, meaning that it does not worsenover time.
The word “nonsyndromic” refers to the fact that there are no other symptoms or systems of the bodyinvolved with the disease. Unlike some other forms of hearing loss, DFNB1 nonsyndromic hearingloss and deafness does not affect balance or movement.
Page 83 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The degree of hearing loss is difficult to predict based on which genetic mutation one has. Even ifmembers of the same family are affected by DFNB1 nonsyndromic hearing loss and deafness, thedegree of hearing loss may vary among them.
How Common is GJB2-related DFNB 1 nonsyndromichearing loss and deafness?In the United States, the United Kingdom, France, Australia, and New Zealand, approximately 14 in100,000 people have DFNB1 nonsyndromic hearing loss and deafness. Roughly 1 in 33 people arecarriers of the mutation that causes the condition.
How is GJB2-related DFNB 1 nonsyndromic hearing lossand deafness Treated?People with DFNB1 nonsyndromic hearing loss and deafness may show improvement by usinghearing aids. For people with profound deafness, cochlear implants may also be helpful. They may alsowant to consider enrolling in an educational program for the hearing impaired.
What is the Prognosis for Someone With GJB2-relatedDFNB 1 nonsyndromic hearing loss and deafness?While a person with GJB2-related DFNB1 nonsyndromic hearing loss and deafness will have mild tosevere hearing loss, it does not affect lifespan and does not affect any other part of the body.
Page 84 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Glucose-6-Phosphate DehydrogenaseDeficiencyGene: G6PD
Variant Accuracy PrecisionV68M 1.000 1.000S188F 1.000 1.000R459P 1.000 1.000R459L 1.000 1.000N126D 1.000 1.000
Detection Rate Population96.74% African American
<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic
74.16% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is glucose-6-phosphate dehydrogenase deficiency?Glucose-6-phosphate dehydrogenase deficiency (G6PD) is an inherited disease in which people arehealthy most of the time, but can develop bouts of fatigue and illness under certain circumstances.These episodes are caused by the destruction of red blood cells (hemolytic anemia), and can be broughton by taking certain types of medication or by the physical stress of a viral or bacterial infection.
It should be emphasized that many people with the mutations that cause this disease do not have anysymptoms.
The symptoms of hemolytic anemia include fatigue, shortness of breath, a yellow tinge to the skin andwhites of the eye (jaundice), rapid heart rate, an enlarged spleen, and overall paleness. In severeepisodes, people can also have brown-colored urine.
Occasionally newborns with the disease show symptoms of jaundice within the first two weeks of life.
Page 85 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Medications known to provoke attacks of hemolytic anemia in people with G6PD include anti-malarialdrugs, common painkillers (such as aspirin or ibuprofen), and certain antibiotics. In people withparticular genetic mutations, contact with fava beans and moth balls—even simply touchingthem—can also provoke attacks. Because of the connection with fava beans, G6PD is sometimesknown as favism.
As the name implies, the disease is caused by a deficiency in an enzyme called glucose-6-phosphatedehydrogenase. A lack of this enzyme can cause red blood cells, which carry oxygen from the lungs tothe rest of the body, to be destroyed faster than the body can replace them.
Please note that G6PD is inherited in an X-linked recessive pattern. Men cannot be carriers of X-linkeddiseases—they either have them or they don't. Because of the way they are inherited, X-linked diseaseslike G6PD are typically more common in men than in women.
How Common is glucose-6-phosphate dehydrogenasedeficiency?G6PD is fairly common, more so in men than in women. Worldwide, G6PD is estimated to affect 400million people. It is common in African populations, with 5 to 25% of males affected depending on thecountry. Roughly 1 in 10 African American males is affected by the disease. G6PD is also common inthe Middle East (notably among Kurds and Sephardic Jews), Asia, and Papua New Guinea.
Researchers believe that G6PD is common because it offers some protection against malaria, aninfectious disease carried by mosquitoes. G6PD is most common in areas where malaria is present.
How is glucose-6-phosphate dehydrogenase deficiencyTreated?Under normal circumstances, people with G6PD do not have any symptoms and require no treatment.They should carefully avoid the medications that can cause bouts of hemolytic anemia. These includeanti-malarial drugs, common painkillers (such as aspirin or ibuprofen), and certain antibiotics. A healthcare provider can provide a complete list of these drugs. People with certain G6PD mutations shouldavoid eating fava beans as well.
If an episode of hemolytic anemia does occur, people with the disease often need bed rest and in severecases, may require blood transfusions.
Once the cause of the episode is resolved (i.e. a drug is discontinued or an infection cleared up),symptoms of the disease usually subside fairly quickly. These episodes often subside without anytreatment.
Page 86 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone Withglucose-6-phosphate dehydrogenase deficiency?The prognosis for a person with G6PD is very good. They typically have no symptoms and can live anormal lifespan with a normal quality of life. In rare cases, episodes of hemolytic anemia can be fatal.Normally people with G6PD who have episodes of hemolytic anemia recover quickly with minimaltreatment.
Page 87 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Glutaric Acidemia Type 1Gene: GCDH
Variant Accuracy PrecisionR402W 1.000 1.000A421V 1.000 1.000
Detection Rate Population13.35% African American13.35% Ashkenazi Jewish13.35% Eastern Asia13.35% Finland13.35% French Canadian or Cajun13.35% Hispanic13.35% Middle East13.35% Native American13.35% Northwestern Europe13.35% Oceania13.35% South Asia13.35% Southeast Asia13.35% Southern Europe
What is glutaric acidemia type 1?Glutaric acidemia type 1 (GA I) is an inherited metabolic disease in which the body lacks an enzyme toproperly break down certain amino acids, the building blocks of protein. When these amino acids buildup in the body, a person can develop brain damage that can impair their ability to move as well as theirintellectual function.
The type and severity of symptoms in people with GA I can vary widely. A small number of peoplewith genetic mutations that cause the disease have no symptoms at all, while others have severemovement problems and/or mental disability.
In most children, symptoms appear between two months and four years of age. A smaller number donot show symptoms until later, even as late as adulthood. Symptoms appear as a “metabolic crisis,” anepisode marked by low blood sugar, vomiting, lack of energy, difficulty feeding, irritability, and poormuscle tone that causes the body to seem floppy. If unrecognized and untreated with a special diet,these episodes can progress to cause spastic and jerking muscle movements, seizures, swelling andbleeding of the brain, coma, and even death. They can often be triggered by illness, fever, or going toolong without eating.
Page 88 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

In children with GA I, the first metabolic crisis often results in permanent damage to a part of the brainknown as the basal ganglia. This damage can result in a severe and permanent loss of motor skills,though often intellect remains normal.
Other children do not experience a metabolic crisis, but show a delay in motor and intellectualdevelopment. Children with these symptoms are more likely to have intellectual impairment later inlife.
Early diagnosis and strict control of the child’s diet can avert a metabolic crisis and significantlyreduce the risk of brain damage and impaired movement ability. Even with careful treatment, however,roughly 25 to 35% of people with GA I develop significant motor problems and other symptoms, evenwithout a metabolic crisis.
Most children with the disease are born with large heads. Some develop bleeding in the brain that hasbeen mistaken for child abuse.
How Common is glutaric acidemia type 1?GA I affects 1 in 40,000 Caucasians. It is more common in certain ethnicities and communities. InSweden, 1 in 30,000 people have the disease. Among Old Order Amish in Pennsylvania and theOjibwa tribe in Canada, as many as 1 in 300 children are affected by GA I.
How is glutaric acidemia type 1 Treated?A physician specializing in metabolism should help devise a treatment plan for any child with GA I.Often these plans include vitamins and supplements and frequent meals low in certain proteins. Dietswill need to be carefully structured to both avoid problem foods and ensure proper nutrition. In somecases, meals may be necessary around the clock, even overnight. A specialist will also devise a “sickday plan” to use when a child shows signs of illness that could lead to a metabolic crisis. Often thisinvolves frequent meals with carbohydrates and an increased fluid intake, even if the child isn't hungryor thirsty.
As children get older, the disease is often easier to manage and the risk of metabolic crises lessens.Many will still need lifelong dietary treatment, however.
It is believed that those who receive treatment before their first metabolic crisis do better in the longterm.
Children who are having a metabolic crisis must be promptly treated, often with intravenous fluids,certain vitamins and supplements, and in some cases, dialysis.
Page 89 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With glutaricacidemia type 1?If treated early and carefully, many children with GA I can live healthy lives with normal or near-normal motor and intellectual development. It should be noted, however, that even with treatment, 25to 35% of children with GA I develop some level of motor and intellectual impairment.
Children who have already had a metabolic crisis are likely to develop permanent brain damage thatcauses severe motor difficulties and involuntary spastic movement. In children who go untreatedduring a metabolic crisis, the disease can be fatal.
Page 90 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
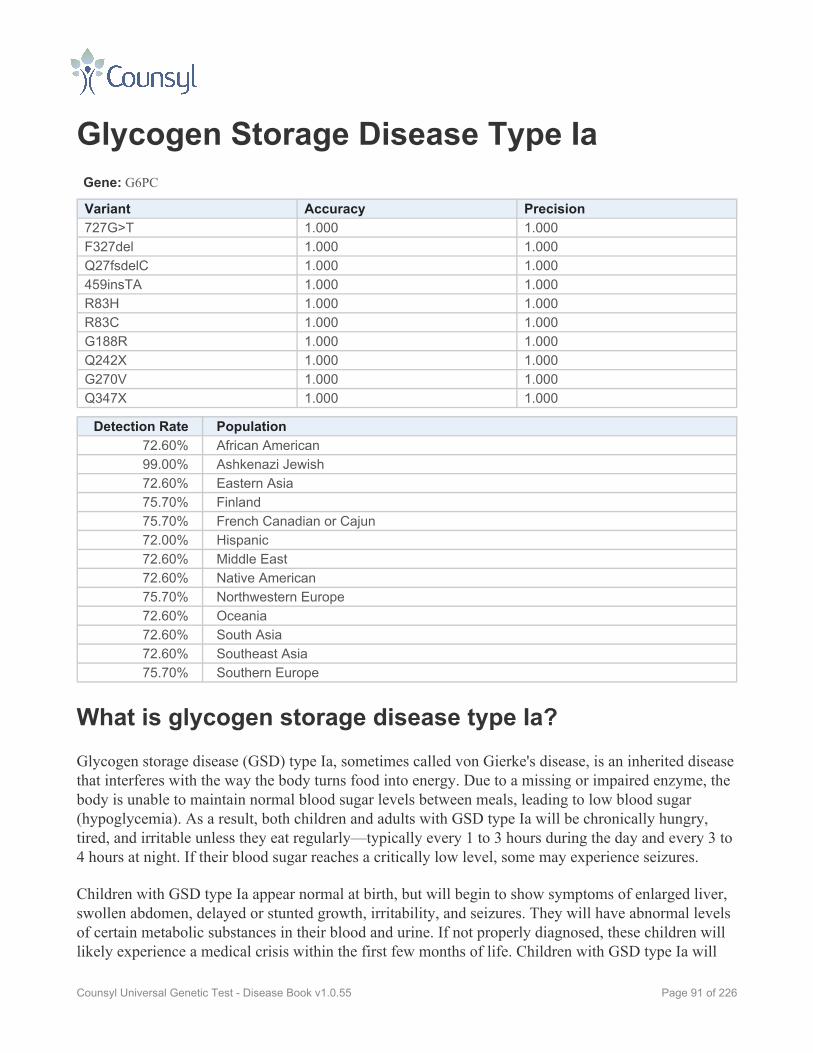
Glycogen Storage Disease Type IaGene: G6PC
Variant Accuracy Precision727G>T 1.000 1.000F327del 1.000 1.000Q27fsdelC 1.000 1.000459insTA 1.000 1.000R83H 1.000 1.000R83C 1.000 1.000G188R 1.000 1.000Q242X 1.000 1.000G270V 1.000 1.000Q347X 1.000 1.000
Detection Rate Population72.60% African American99.00% Ashkenazi Jewish72.60% Eastern Asia75.70% Finland75.70% French Canadian or Cajun72.00% Hispanic72.60% Middle East72.60% Native American75.70% Northwestern Europe72.60% Oceania72.60% South Asia72.60% Southeast Asia75.70% Southern Europe
What is glycogen storage disease type Ia?Glycogen storage disease (GSD) type Ia, sometimes called von Gierke's disease, is an inherited diseasethat interferes with the way the body turns food into energy. Due to a missing or impaired enzyme, thebody is unable to maintain normal blood sugar levels between meals, leading to low blood sugar(hypoglycemia). As a result, both children and adults with GSD type Ia will be chronically hungry,tired, and irritable unless they eat regularly—typically every 1 to 3 hours during the day and every 3 to4 hours at night. If their blood sugar reaches a critically low level, some may experience seizures.
Children with GSD type Ia appear normal at birth, but will begin to show symptoms of enlarged liver,swollen abdomen, delayed or stunted growth, irritability, and seizures. They will have abnormal levelsof certain metabolic substances in their blood and urine. If not properly diagnosed, these children willlikely experience a medical crisis within the first few months of life. Children with GSD type Ia will
Page 91 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

typically be shorter than their family members and will experience delayed puberty. Mental function isnot affected by GSD type Ia.
These symptoms occur due to the lack or improper functioning of an enzyme calledglucose-6-phosphatase (G6Pase). Normally, glucose (a sugar) in the food we eat is converted into asubstance called glycogen that is stored in the liver. When a person does not eat for 3 to 4 hours, thisglycogen will be turned back into glucose and used to stabilize sugar levels in the body. People withGSD Ia lack a component of G6Pase so they cannot turn glycogen back into glucose, and thereforethey suffer from hypoglycemia.
People with GSD type Ib lack a different component of G6Pase, and experience similar symptoms. Forthis reason, type Ia and Ib are often spoken about as one disease: GSD type I.
The buildup of glycogen in the liver and kidneys causes them to swell, although these organs are stillable to perform the majority of their functions. Benign (non-cancerous) tumors in the liver are oftenseen around the time of puberty. Rarely, these can become cancerous. Changes in kidney function mayoccur as the person reaches his or her 20s, and may include kidney stones and a decreased ability tofilter waste products. In advanced cases, dialysis and/or a kidney transplant may be needed.
How Common is glycogen storage disease type Ia?Roughly 1 in every 20,000 to 25,000 babies in the U.S. and Europe is born with some form ofGlycogen Storage Disease. Of those, 25% have type I (either Ia or Ib).
How is glycogen storage disease type Ia Treated?The treatment of GSD type Ia involves a careful monitoring of the affected person’s diet, both infrequency of meals and type of foods eaten. People with GSD type Ia should avoid foods with sucrose(table sugar), fructose (sugar from fruits), and lactose and galactose (sugars found in milk). They needto eat around the clock, typically every 1 to 3 hours during the day and every 3 to 4 hours at night, tomaintain healthy blood sugar levels.
Infants and young children often need a feeding tube in order to tolerate frequent eating. They mayalso need to use a feeding pump at night and for emergency feedings should their blood sugar dropdangerously low. Because they must eat so frequently, children with GSD type Ia often have problemseating and swallowing food orally. They often need therapy to re-learn sucking, swallowing, and evenspeech.
Physicians often recommend people with GSD type Ia drink cornstarch mixed with water, soy formula,or soy milk. Cornstarch is digested slowly and therefore releases its glucose gradually, helping tosafely extend the time between meals.
Page 92 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With glycogenstorage disease type Ia?With careful monitoring of diet and blood sugar levels, people with GSD can cope with their metabolicabnormalities and live into adulthood. Without close monitoring of the diet, extremely low blood sugarcan be fatal. In adolescence and adulthood, people with the disease must be alert to kidneycomplications, high blood pressure, and/or cancerous liver tumors.
Page 93 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Glycogen Storage Disease Type IbGene: G6PT1
Variant Accuracy Precision1211delCT 1.000 1.000G339C 1.000 1.000G339D 1.000 1.000A367T 1.000 1.000
Detection Rate Population<10% African American
48.41% Ashkenazi Jewish<10% Eastern Asia
48.41% Finland48.41% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
48.41% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
48.41% Southern Europe
What is glycogen storage disease type Ib?Glycogen storage disease (GSD) type Ib is an inherited disease that interferes with the way the bodyturns food into energy. Due to a missing or impaired enzyme, the body is unable to maintain normalblood sugar levels between meals, leading to low blood sugar (hypoglycemia). As a result, bothchildren and adults with GSD type Ib will be chronically hungry, tired, and irritable unless they eatregularly—typically every 1 to 3 hours during the day and every 3 to 4 hours at night. If their bloodsugar reaches a critically low level, some may experience seizures.
Children with GSD type Ib appear normal at birth, but will begin to show symptoms of enlarged liver,swollen abdomen, delayed or stunted growth, irritability, and seizures. They will have abnormal levelsof certain metabolic substances in their blood and urine. If not properly diagnosed, these children willlikely experience a medical crisis within the first few months of life. Children with GSD type Ib willtypically be shorter than their family members and will experience delayed puberty. Mental function isnot affected by GSD type Ib.
Individuals with GSD type Ib are also prone to frequent bacterial and fungal infections, due toabnormalities in and/or low levels of a type of white blood cell called neutrophils. They are also more
Page 94 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

likely to develop chronic inflammation of the pancreas, chronic inflammatory bowel disease, andCrohn's disease.
The symptoms of GSD type Ib occur due to the lack or improper functioning of an enzyme calledglucose-6-phosphatase (G6Pase). Normally, glucose (a sugar) in the food we eat is converted into asubstance called glycogen that is stored in the liver. When a person does not eat for 3 to 4 hours, thisglycogen will be turned back into glucose and used to stabilize sugar levels in the body. People withGSD Ib lack a component of G6Pase so they cannot turn glycogen back into glucose, and thereforethey suffer from hypoglycemia.
People with GSD type Ia lack a different component of G6Pase, and experience similar symptoms. Forthis reason, type Ia and Ib are often spoken about as one disease: GSD type I.
The buildup of glycogen in the liver and kidneys causes them to swell, although these organs are stillable to perform the majority of their functions. Benign (non-cancerous) tumors in the liver are oftenseen around the time of puberty. Rarely, these can become cancerous. Changes in kidney function mayoccur as the person reaches his or her 20s, and may include kidney stones and a decreased ability tofilter waste products. In people with advanced cases, dialysis and/or a kidney transplant may beneeded.
How Common is glycogen storage disease type Ib?Roughly 1 in every 20,000 to 25,000 babies in the U.S. and Europe is born with some form ofGlycogen Storage Disease. Of those, 25% have type I (either Ia or Ib).
How is glycogen storage disease type Ib Treated?The treatment of GSD type Ib involves a careful monitoring of the affected person's diet, both infrequency of meals and type of foods eaten. People with GSD type Ib should avoid foods with sucrose(table sugar), fructose (sugar from fruits), and lactose and galactose (sugars found in milk). They needto eat around the clock, typically every 1 to 3 hours during the day and every 3 to 4 hours at night, tomaintain healthy blood sugar levels.
Infants and young children often need a feeding tube in order to tolerate frequent eating. They mayalso need to use a feeding pump at night and for emergency feedings should their blood sugar dropdangerously low. Because they must eat so frequently, children with GSD type Ib often have problemseating and swallowing food orally. They often need therapy to re-learn sucking, swallowing, and evenspeech.
Physicians often recommend people with GSD type Ib drink cornstarch mixed with water, soy formula,or soy milk. Cornstarch is digested slowly and therefore releases its glucose gradually, helping tosafely extend the time between meals.
Page 95 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

People affected by GSD type Ib also frequently take medication to increase the number of neutrophils,a type of white blood cell that fights infection. They must also be vigilant to treat any infection in thebody as it arises.
What is the Prognosis for Someone With glycogenstorage disease type Ib?With careful monitoring of diet and blood sugar levels, people with GSD type Ib can improve theirmetabolic abnormalities and live into adulthood. Without close monitoring of the diet however,extremely low blood sugar can be fatal. In adolescence and adulthood, people with GSD type Ib mustbe alert to infections, kidney complications, high blood pressure, and/or cancerous liver tumors. Long-term complications can include kidney damage, brittle bones (osteoporosis), benign cysts on theovaries (in women), and benign tumors of the liver (adenomas).
Page 96 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Glycogen Storage Disease Type IIIGene: AGL
Variant Accuracy PrecisionQ6X 1.000 1.00017delAG 1.000 1.0001484delT 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is glycogen storage disease type III?Glycogen storage disease type III (GSD III)—also known as Cori disease or Forbes disease—is aninherited condition in which the deficiency of an enzyme called glycogen debranching enzyme resultsin various complications, notably involving the liver and muscles. The lack of this enzyme means thebody cannot properly break down glycogen, a stored form of sugar. As a result, glycogen cannotproperly be used to energize the body and glycogen molecules accumulate in the body.
Symptoms of GSD III often appear in infancy or childhood. The liver is enlarged, leading to anoticeably swollen abdomen. This enlargement usually subsides with puberty, although there may belong term liver damage.
Muscle weakness is a problem seen in 85% of people with GSD III. This weakness can at times besevere and may worsen in adulthood. Children with the disease may experience delayed growth, butusually reach normal adult height. A minority of people with the disease also have a mildly enlargedheart, though its function is usually normal.
Low blood sugar and feelings of tiredness may occur with GSD III, although they are less common andless severe than with another form of GSD, type I.
Page 97 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The onset of the disease may occur in adulthood, which typically corresponds with milder symptoms.
How Common is glycogen storage disease type III?About 1 in 100,000 U.S. births is affected by GSD III. This disease is much more common in IsraeliJews of North African descent, where 1 in 35 are carriers and 1 in 5,400 babies has the disease. Thehighest rate is found among people on the Faroe Islands of the North Atlantic, where 1 in 30 is acarrier and 1 in 3,600 babies is affected.
How is glycogen storage disease type III Treated?There is no treatment for the cause of GSD III. Physicians will monitor the liver, heart, and muscles inaffected people and recommend physical therapy when necessary to promote better movement.Frequent small meals and a high-protein diet may also be beneficial. Physicians may recommendconsuming corn starch, which breaks down slowly into simple sugars and may alleviate symptoms oflow blood sugar between meals. Parents of infants should be particularly careful to monitor the child’sdiet to avoid hypoglycemic seizures.
What is the Prognosis for Someone With glycogenstorage disease type III?Among infants with GSD III, there is an increased rate of fatalities due to seizures caused by low bloodsugar. While an exact lifespan is unknown, many people with GSD III live well into adulthood. Liverdisease and muscle weakness may contribute to a cause of death long-term.
Page 98 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Glycogen Storage Disease Type VGene: PYGM
Variant Accuracy PrecisionR49X 1.000 1.000G204S 1.000 1.000K542T 1.000 1.000K542X 1.000 1.000
Detection Rate Population<10% African American
68.83% Ashkenazi Jewish<10% Eastern Asia
68.83% Finland68.83% French Canadian or Cajun65.83% Hispanic
<10% Middle East<10% Native American
68.83% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
65.83% Southern Europe
What is glycogen storage disease type V?Glycogen storage disease type V (GSD-V), also called McArdle disease, is an inherited disease inwhich the body lacks an enzyme called myophosphorylase. This deficiency prevents an affected personfrom turning glycogen, a stored form of sugar, into glucose, which can be used for energy. People withGSD-V have difficulty doing exercise of any kind because the body lacks its normal source of energy,glucose, and instead breaks down its own muscles. For people with GSD-V, exercise will cause pain,cramping, fatigue, and soreness to the muscle tissue. Some people with GSD-V find that after an initialdifficulty with exercise, they get a “second wind” and are able to continue. Muscle strength is oftennormal.
In half of people with GSD-V, the breakdown of muscle also causes the release of a protein calledmyoglobin into the bloodstream, resulting in dark red urine. If a person with GSD-V exercisesexcessively, large amounts of myoglobin may overwhelm the kidneys and cause temporary kidneyfailure.
Symptoms of GSD-V usually begin in one's 20s or 30s, but can begin anywhere from infancy to lateradulthood. The severity of the symptoms can vary, but in many people symptoms do not get
Page 99 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

significantly worse over time. About a third of adults with the disease experience progressive muscleweakening later in life. The degree of enzyme deficiency can determine the severity of the disease.
How Common is glycogen storage disease type V?GSD-V is rare, but its exact frequency is unknown. One study showed the disease incidence to be 1 in100,000 in the Dallas-Fort Worth area of Texas in the United States.
How is glycogen storage disease type V Treated?There is no treatment for GSD-V other than to restrict the frequency and intensity of exercise. Toomuch physical exertion will result in muscle breakdown and severe kidney problems. The results froma study of 12 people affected by GSD-V suggested that eating the sugar sucrose before exercise mayminimize muscle pain and injury.
What is the Prognosis for Someone With glycogenstorage disease type V?If people with GSD-V properly manage their physical activity, the disease should not significantlyimpact their lifespan. Those who exercise only moderately can lead normal lives.
Page 100 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

GRACILE SyndromeGene: BCS1L
Variant Accuracy PrecisionS78G 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia>99% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is GRACILE syndrome?GRACILE syndrome is a fatal inherited disorder caused by a mutation in a gene necessary forproviding cells with energy. “GRACILE” is an acronym for symptoms caused by the disease:
Growth Retardation The baby is born significantly smaller than normal.
Aminoaciduria There are increased levels of amino acids in the child’s urine.
Cholestasis The flow of bile from the liver is blocked.
Iron overload The body does not metabolize iron properly, leading to excess levels, particularly inthe liver.
Lactic acidosis Lactic acid builds up in the body, causing the blood to become too acidic.
Early death Half of babies with GRACILE syndrome die within 24 hours. Virtually none live beyondthe age of four months.
Page 101 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is GRACILE syndrome?GRACILE syndrome has only been reported in Finland. Here, researchers estimate that 1 in 47,000babies are affected, meaning that roughly 1 in 110 Finnish people are carriers of the genetic mutation.There have been reports of several infants in the United Kingdom and Turkey with similar but notidentical symptoms. It is not known whether this represents the same disease.
How is GRACILE syndrome Treated?There is no effective treatment for GRACILE syndrome. Experimental treatments to reduce acidityand/or iron levels in the infants’ blood have not been shown to extend life beyond several months.
What is the Prognosis for Someone With GRACILEsyndrome?About half of infants with GRACILE syndrome die within the first few days of life. Even with the bestpossible treatment, nearly all will die within four months.
Page 102 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hereditary Fructose IntoleranceGene: ALDOB
Variant Accuracy PrecisionDelta4E4 1.000 1.000Y204X 1.000 1.000A149P 1.000 1.000N334K 1.000 1.000
Detection Rate Population<10% African American
67.82% Ashkenazi Jewish<10% Eastern Asia
67.82% Finland67.82% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
67.82% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
67.82% Southern Europe
What is hereditary fructose intolerance?Hereditary fructose intolerance (HFI) is a condition in which the body lacks a substance called aldolaseB needed to process fructose, a common sugar found in fruit and many other foods. When a personwith HFI consumes fructose, the result is low blood sugar (hypoglycemia) and a buildup of toxicsubstances in the liver.
Infants or children with the disease who consume the sugars fructose and sucrose or the sugarsubstitute sorbitol typically experience symptoms after eating, including vomiting, convulsions,irritability, and/or sleepiness. Many infant formulas are made with the sugar lactose, although somealso contain fructose and sucrose, as do many baby foods. Infants or children with HFI may show ayellowing of the skin and whites of the eyes (jaundice) and have an enlarged liver and spleen. Ifunrecognized and untreated, these children will fail to grow at a normal rate.
If the disease is not detected and treated, HFI can lead to serious liver disease, hypoglycemic shock,seizures, gout, bleeding, and kidney or liver failure. In extreme cases, it can be fatal. For this reason,early detection is critical.
Page 103 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

A strict diet free from fructose, sucrose, and sorbitol allows most people with HFI to live normal,symptom-free lives, although those with a severe course of the disease may develop serious liverdisease later in life, even with a careful diet.
Symptoms of the disease can vary from mild to severe. People with HFI often show an aversion tosweets and fruit, and thus those with a mild case may be protected from some of the symptoms theywould otherwise experience.
How Common is hereditary fructose intolerance?The exact prevalence of HFI is unknown, but several studies have placed the number of affectedpeople in the U.S. and Europe at 1 in 20,000. A recent U.K. study placed the figure at 1 in 12,000 to58,000.
How is hereditary fructose intolerance Treated?The key to treating people with HFI is to strictly control their diet, eliminating all fructose, sucrose,and sorbitol. On this careful diet, people with HFI can be symptom-free, though symptoms will quicklyreturn upon consuming fructose, sucrose, or sorbitol.
In cases where liver disease has progressed to a life-threatening stage, liver transplantation is a possibletreatment.
What is the Prognosis for Someone With hereditaryfructose intolerance?Without a careful monitoring of the diet—elimination of all fructose, sucrose, and sorbitol—HFI canbe life-threatening, causing serious liver disease, hypoglycemic shock, or liver or kidney failure.
With a careful diet, however, people with HFI may be symptom free and able to live normal lives. Theearlier the condition is diagnosed and the diet corrected, the less damage is done to the liver andkidneys and the better the overall prognosis. Early detection and diet modification is also important sothat children can grow to normal height. Within three to four weeks of adopting a fructose-free diet,people with HFI can be symptom-free.
In a minority of people who have a severe form of the disease, liver disease may still develop, despite acareful diet.
Page 104 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hereditary Thymine-UraciluriaGene: DPYD
Variant Accuracy PrecisionIVS14+1G>A 1.000 1.000
Detection Rate Population<10% African American
52.00% Ashkenazi Jewish<10% Eastern Asia
52.00% Finland52.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
52.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
52.00% Southern Europe
What is hereditary thymine-uraciluria?Hereditary thymine-uraciluria is an inherited disease that can cause serious mental and physical delaysin children. For reasons that are not understood, most people with the genetic mutations that causehereditary thymine-uraciluria have no symptoms at any time in their lives, while others are severelyaffected in infancy or childhood.
Among those who are affected, about 50% have neurological symptoms including seizures, mentaldisability, and a delay in motor skills. Less common symptoms include autism, a small head, a delay inphysical growth, eye abnormalities, and speech difficulties. These symptoms typically appear ininfancy or childhood.
All people with hereditary thymine-uraciluria, regardless of the presence or absence of symptoms,cannot properly break down the common chemotherapy drug 5-fluorouracil. If given this drug, theywill have a severe toxic reaction that could be life-threatening. Signs of this reaction include diarrhea,swelling, digestive problems, muscle weakness, and an inability to coordinate muscle movement.Carriers of a mutation in the gene that causes this disease are also at risk for toxicity following5-fluorouracil treatment.
Hereditary thymine-uraciluria is caused by the absence of an enzyme called dihydropyrimidinedehydrogenase which is needed for breaking down the molecules thymine and uracil, and also5-fluorouracil when it is present in the body.
Page 105 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is hereditary thymine-uraciluria?Studies have shown that about 1% of Caucasians are carriers for a particular mutation that causeshereditary thymine-uraciluria. This mutation is common among people in the Netherlands, Finland,and Taiwan. Due to this mutation and other mutations in the same gene, an estimated 3% ofCaucasians and 8% of African Americans are at risk for 5-fluorouracil toxicity.
How is hereditary thymine-uraciluria Treated?There is no cure for hereditary thymine-uraciluria. Its symptoms can only be addressed as they arise(i.e. medication to prevent seizures). People with this disease must not take the drug 5-fluorouracil inorder to avoid a toxic reaction.
What is the Prognosis for Someone With hereditarythymine-uraciluria?For those who are asymptomatic, the prognosis is very good. Their lifespan should be unaffected bythe disease. For those with more severe symptoms, it is unknown how these symptoms affect lifespan.
Page 106 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Herlitz Junctional Epidermolysis Bullosa,LAMA3-RelatedGene: LAMA3
Variant Accuracy PrecisionR650X 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Herlitz junctional epidermolysis bullosa,LAMA3-related?Herlitz junctional epidermolysis bullosa (H-JEB) is an inherited disease that causes severe blisteringon the skin. Infants with H-JEB frequently also have internal blistering on the lining of the nose,mouth, esophagus, trachea, rectum, stomach, intestines, and eyes. These symptoms are present frombirth. Occasionally people with the disease survive into their teens, however 87% die in the first yearof life.
People with H-JEB lack anchors to hold the layers of their skin together. They develop large, fluid-filled blisters in response to any trauma, even something as minor as increased room temperature. Skinchafes and wears away, leaving the person open to infection.
Granulation tissue, a kind of soft, pink, bumpy, moist skin, is often seen around the nose, mouth, ears,fingers, and toes, as well as in areas that receive friction, such as the buttocks and back of the head.This tissue bleeds easily and can be a site of fluid loss.
Infants and children with the disease often develop a hoarse cry, cough, and other breathing problems.They are prone to developing fevers, often lose their fingernails and toenails, and have poorly-formed
Page 107 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

tooth enamel. They may also have abnormalities in their urinary tract and bladder which can lead tourinary tract infections and kidney failure.
These infants do not grow at the expected rate and may also develop electrolyte imbalances, hair loss,osteoporosis, and skin cancer.
How Common is Herlitz junctional epidermolysis bullosa,LAMA3-related?H-JEB is extremely rare. Estimates indicate that 0.37 people per million are affected by the disease and1 in 781 Americans is a carrier.
How is Herlitz junctional epidermolysis bullosa,LAMA3-related Treated?Even with the best of care, H-JEB is ultimately fatal. There are no successful treatments other than toprotect the child as much as possible from skin damage and treat symptoms as they arise. A cesareansection may be recommended to protect the child from the skin trauma of birth.
Open wounds and blistered skin is often covered with multiple layers of non-adhesive bandages andanyone handling the child must use extreme care. The child must avoid any movement or clothing thatcould damage the skin.
Antibiotics are often prescribed for infection and antiseptics used to prevent infection. A dietitianshould be consulted for an infant’s proper nutrition. People with H-JEB should drink plenty of fluids toavoid dehydration.
To aid in breathing, an opening may be made in the neck to deliver air to the trachea, however this maybe difficult on a person with fragile skin.
What is the Prognosis for Someone With Herlitzjunctional epidermolysis bullosa, LAMA3-related?The prognosis for a person with H-JEB is poor. Roughly 87% will die within the first year of life, andall will die by the late teens. The disease is extremely painful. Causes of death often include infection,breathing problems, and loss of fluid leading to dehydration.
Page 108 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Herlitz Junctional Epidermolysis Bullosa,LAMB3-RelatedGene: LAMB3
Variant Accuracy Precision3024delT 1.000 1.000R42X 1.000 1.000R144X 1.000 1.000Q243X 1.000 1.000R635X 1.000 1.000
Detection Rate Population51.69% African American51.69% Ashkenazi Jewish51.69% Eastern Asia51.69% Finland51.69% French Canadian or Cajun51.69% Hispanic51.69% Middle East51.69% Native American51.69% Northwestern Europe51.69% Oceania51.69% South Asia51.69% Southeast Asia51.69% Southern Europe
What is Herlitz junctional epidermolysis bullosa,LAMB3-related?Herlitz junctional epidermolysis bullosa (H-JEB) is an inherited disease that causes severe blisteringon the skin. Infants with H-JEB frequently also have internal blistering on the lining of the nose,mouth, esophagus, trachea, rectum, stomach, intestines, and eyes. These symptoms are present frombirth. Occasionally people with the disease survive into their teens, however 87% die in the first yearof life.
People with H-JEB lack anchors to hold the layers of their skin together. They develop large, fluid-filled blisters in response to any trauma, even something as minor as increased room temperature. Skinchafes and wears away, leaving the person open to infection.
Granulation tissue, a kind of soft, pink, bumpy, moist skin, is often seen around the nose, mouth, ears,fingers, and toes, as well as in areas that receive friction, such as the buttocks and back of the head.This tissue bleeds easily and can be a site of fluid loss.
Page 109 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Infants and children with the disease often develop a hoarse cry, cough, and other breathing problems.They are prone to developing fevers, often lose their fingernails and toenails, and have poorly-formedtooth enamel. They may also have abnormalities in their urinary tract and bladder which can lead tourinary tract infections and kidney failure.
These infants do not grow at the expected rate and may also develop electrolyte imbalances, hair loss,osteoporosis, and skin cancer.
How Common is Herlitz junctional epidermolysis bullosa,LAMB3-related?H-JEB is extremely rare. Estimates indicate that 0.37 people per million are affected by the disease and1 in 781 Americans is a carrier.
How is Herlitz junctional epidermolysis bullosa,LAMB3-related Treated?Even with the best of care, H-JEB is ultimately fatal. There are no successful treatments other than toprotect the child as much as possible from skin damage and treat symptoms as they arise. A cesareansection may be recommended to protect the child from the skin trauma of birth.
Open wounds and blistered skin is often covered with multiple layers of non-adhesive bandages andanyone handling the child must use extreme care. The child must avoid any movement or clothing thatcould damage the skin.
Antibiotics are often prescribed for infection and antiseptics used to prevent infection. A dietitianshould be consulted for an infant’s proper nutrition. People with H-JEB should drink plenty of fluids toavoid dehydration.
To aid in breathing, an opening may be made in the neck to deliver air to the trachea, however this maybe difficult on a person with fragile skin.
What is the Prognosis for Someone With Herlitzjunctional epidermolysis bullosa, LAMB3-related?The prognosis for a person with H-JEB is poor. Roughly 87% will die within the first year of life, andall will die by the late teens. The disease is extremely painful. Causes of death often include infection,breathing problems, and loss of fluid leading to dehydration.
Page 110 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Herlitz Junctional Epidermolysis Bullosa,LAMC2-RelatedGene: LAMC2
Variant Accuracy PrecisionR95X 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Herlitz junctional epidermolysis bullosa,LAMC2-related?Herlitz junctional epidermolysis bullosa (H-JEB) is an inherited disease that causes severe blisteringon the skin. Infants with H-JEB frequently also have internal blistering on the lining of the nose,mouth, esophagus, trachea, rectum, stomach, intestines, and eyes. These symptoms are present frombirth. Occasionally people with the disease survive into their teens, however 87% die in the first yearof life.
People with H-JEB lack anchors to hold the layers of their skin together. They develop large, fluid-filled blisters in response to any trauma, even something as minor as increased room temperature. Skinchafes and wears away, leaving the person open to infection.
Granulation tissue, a kind of soft, pink, bumpy, moist skin, is often seen around the nose, mouth, ears,fingers, and toes, as well as in areas that receive friction, such as the buttocks and back of the head.This tissue bleeds easily and can be a site of fluid loss.
Infants and children with the disease often develop a hoarse cry, cough, and other breathing problems.They are prone to developing fevers, often lose their fingernails and toenails, and have poorly-formed
Page 111 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

tooth enamel. They may also have abnormalities in their urinary tract and bladder which can lead tourinary tract infections and kidney failure.
These infants do not grow at the expected rate and may also develop electrolyte imbalances, hair loss,osteoporosis, and skin cancer.
How Common is Herlitz junctional epidermolysis bullosa,LAMC2-related?H-JEB is extremely rare. Estimates indicate that 0.37 people per million are affected by the disease and1 in 781 Americans is a carrier.
How is Herlitz junctional epidermolysis bullosa,LAMC2-related Treated?Even with the best of care, H-JEB is ultimately fatal. There are no successful treatments other than toprotect the child as much as possible from skin damage and treat symptoms as they arise. A cesareansection may be recommended to protect the child from the skin trauma of birth.
Open wounds and blistered skin is often covered with multiple layers of non-adhesive bandages andanyone handling the child must use extreme care. The child must avoid any movement or clothing thatcould damage the skin.
Antibiotics are often prescribed for infection and antiseptics used to prevent infection. A dietitianshould be consulted for an infant’s proper nutrition. People with H-JEB should drink plenty of fluids toavoid dehydration.
To aid in breathing, an opening may be made in the neck to deliver air to the trachea, however this maybe difficult on a person with fragile skin.
What is the Prognosis for Someone With Herlitzjunctional epidermolysis bullosa, LAMC2-related?The prognosis for a person with H-JEB is poor. Roughly 87% will die within the first year of life, andall will die by the late teens. The disease is extremely painful. Causes of death often include infection,breathing problems, and loss of fluid leading to dehydration.
Page 112 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
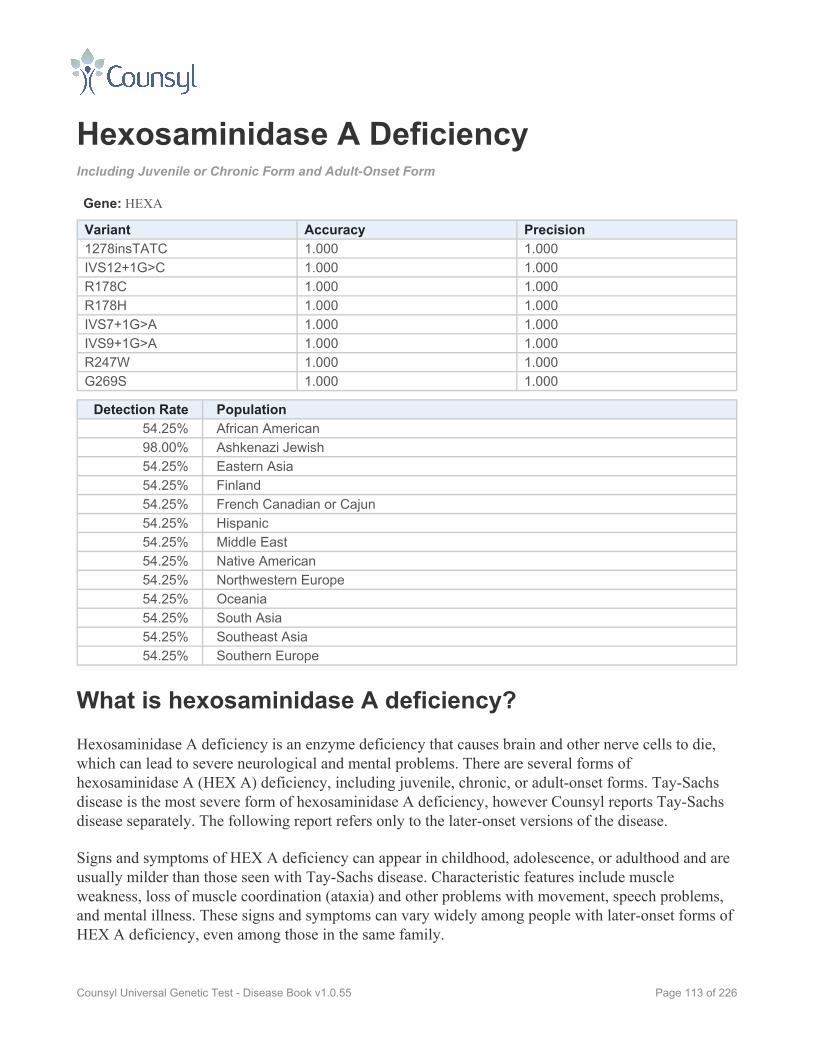
Hexosaminidase A DeficiencyIncluding Juvenile or Chronic Form and Adult-Onset Form
Gene: HEXA
Variant Accuracy Precision1278insTATC 1.000 1.000IVS12+1G>C 1.000 1.000R178C 1.000 1.000R178H 1.000 1.000IVS7+1G>A 1.000 1.000IVS9+1G>A 1.000 1.000R247W 1.000 1.000G269S 1.000 1.000
Detection Rate Population54.25% African American98.00% Ashkenazi Jewish54.25% Eastern Asia54.25% Finland54.25% French Canadian or Cajun54.25% Hispanic54.25% Middle East54.25% Native American54.25% Northwestern Europe54.25% Oceania54.25% South Asia54.25% Southeast Asia54.25% Southern Europe
What is hexosaminidase A deficiency?Hexosaminidase A deficiency is an enzyme deficiency that causes brain and other nerve cells to die,which can lead to severe neurological and mental problems. There are several forms ofhexosaminidase A (HEX A) deficiency, including juvenile, chronic, or adult-onset forms. Tay-Sachsdisease is the most severe form of hexosaminidase A deficiency, however Counsyl reports Tay-Sachsdisease separately. The following report refers only to the later-onset versions of the disease.
Signs and symptoms of HEX A deficiency can appear in childhood, adolescence, or adulthood and areusually milder than those seen with Tay-Sachs disease. Characteristic features include muscleweakness, loss of muscle coordination (ataxia) and other problems with movement, speech problems,and mental illness. These signs and symptoms can vary widely among people with later-onset forms ofHEX A deficiency, even among those in the same family.
Page 113 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Those with juvenile onset HEX A deficiency typically begin to show symptoms between the ages of 2and 10. Early signs can include a decline in verbal skills, the ability to accomplish life skills, andoverall thought processes. These symptoms become progressively worse over time. The child will alsodevelop spastic movement, seizures, and vision loss. Most children with HEX A deficiency will enter avegetative state by the age of 10 to 15.
Similarly, symptoms of the chronic form of the disease can begin any time from early childhood untilthe age of 10. While this form of HEX A deficiency can cause a variety of movement problems, thedecline of verbal skills and thought processes tends to happen later than in the juvenile form.
Adult-onset HEX A deficiency is rare, but it is thought that symptoms often begin in one's 20s or 30s.This form of the disease has the biggest variation in symptoms from person to person, even amongpeople in the same family. Often its symptoms appear identical to other more common diseases,leading to misdiagnoses. These symptoms can include some or all of the following: muscle weakness,involuntary muscle twitching, speech difficulties, altered thought, or severe mental disorders likepsychosis or schizophrenia. Some people with adult-onset HEX A deficiency may develop movementdisorders but be spared mental decline until their 60s or 70s. For some, adult-onset HEX A deficiencyis not fatal.
HEX A deficiency is caused by the near absence of an enzyme called beta-hexosaminidase A. Thisenzyme helps break down a particular fatty acid called GM2 ganglioside. Without adequate amounts offunctional enzymes, GM2 ganglioside will build up in nerve cells and cause them to die.
How Common is hexosaminidase A deficiency?Tay-Sachs Disease is by far the most common form of hexosaminidase A deficiency. The other formsof the disease discussed here are extremely rare. Their exact prevalence is unknown. Mutations thatcause these later-onset forms have been found in multiple ethnic groups.
How is hexosaminidase A deficiency Treated?At this time there is no cure for hexosaminidase A deficiency. Treatment largely addresses symptomsas they arise, such as aiding mobility with mechanical aids or controlling seizures and mental disorderswith medication. Because the symptoms of these forms of the disease vary widely, treatment isdependent upon the type of symptoms and their severity.
What is the Prognosis for Someone With hexosaminidaseA deficiency?The prognosis for a person with hexosaminidase A deficiency can vary widely, depending on when inlife the symptoms begin and the severity of the individual's case.
Page 114 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Those with juvenile or chronic HEX A deficiency typically experience severe mental decline betweenthe ages of 2 and 10 and often reach a vegetative state between the ages of 10 to 15, with deathfollowing several years later. In more severe cases, someone with juvenile HEX A deficiency can diein early childhood.
Those with the adult-onset form of the disease face more varied outcomes. Some may develop severemental problems such as psychosis by age 20 while others may reach their 60s or 70s with movementdifficulty, but without mental problems. The lifespan of people with adult-onset HEX A deficiency arenot well understood and can be difficult to predict. In some cases, the disease has not impactedlifespan.
Page 115 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

HFE-Associated HereditaryHemochromatosisGene: HFE
Variant Accuracy PrecisionH63D 1.000 1.000S65C 1.000 1.000Q127H 1.000 1.000E168Q 1.000 1.000E168X 1.000 1.000W169X 1.000 1.000C282Y 1.000 1.000Q283P 1.000 1.000V53M 1.000 1.000V59M 1.000 1.000H63H 1.000 1.000
Detection Rate Population<10% African American
90.00% Ashkenazi Jewish<10% Eastern Asia
90.00% Finland90.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
90.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
90.00% Southern Europe
What is HFE-associated hereditary hemochromatosis?HFE-associated hereditary hemochromatosis (HFE-HHC) is a common and treatable inherited diseasein which the body absorbs and stores too much iron, potentially damaging organs such as the liver,heart, and pancreas. If the disease is diagnosed and treated before symptoms develop, people withHFE-HHC typically have a normal lifespan. If the disease is untreated, however, it can lead to fatalliver and heart failure.
For reasons not well understood, the majority of people with the genetic mutations that cause HFE-HHC do not develop symptoms of the disease at any point in their lives. For these people, simple blood
Page 116 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

tests can determine whether or not the body is storing too much iron. If it is, beginning treatment earlycan leave a person virtually symptom-free for life.
Depending on the specific mutation(s) a person has, he or she can be more or less likely to develop theiron overload symptoms of HFE-HHC.
People who have two copies of the C282Y mutation are most likely to have dangerously elevatedlevels of iron in their blood. Studies have found that among those with the C282Y/C282Ycombination, men are more likely to develop symptoms of iron overload than women, perhaps becausewomen's menstrual cycles lower their iron levels on a regular basis. Do keep in mind, however, that themajority of people with two copies of the C282Y mutation do not develop any symptoms of HFE-HHC.
Those who have C282Y in combination with another HFE-HHC mutation are much less likely todevelop symptoms of the disease. Only 0.5% to 2% of people with C282Y in combination with anothermutation are thought to have clinical signs of the disease. People with this genetic combination whohave another disease of the liver may be more likely to develop HFE-HHC symptoms.
Among people who have two copies of any other HFE-HHC mutation, including a very commonmutation known as H63D, the likelihood of developing symptoms is extremely low. In the absence ofanother liver disease, two copies of any HFE-HHC mutation other than C282Y is unlikely to cause anyhealth problems.
In men who have not been treated for HFE-HHC, the first symptoms of the disease typically beginbetween the ages of 30 to 50; for untreated women, symptoms usually begin later, after menopause.
Early symptoms often include weakness, abdominal pain, joint pain, weight loss, loss of interest in sex,chest pain, and a progressive gray or bronze pigmentation to the skin. Liver disease (either fibrosis orthe more serious cirrhosis) is a common problem associated with HFE-HHC. Cirrhosis can lead to fatalliver failure and/or an increased likelihood of developing cancer of the liver.
The heart can also be affected by HFE-HHC, seen as an irregular heartbeat and/or congestive heartfailure. Other problems caused by HFE-HHC can include diabetes, arthritis, impotence (in men), earlymenopause (in women), thyroid problems, and adrenal gland problems.
How Common is HFE-associated hereditaryhemochromatosis?HFE-HHC mutations are extremely common, particularly among Caucasians. Roughly one-third(33%) of Caucasians are carriers of the condition, most commonly the H63D mutation. The H63Dmutation is almost always associated with asymptomatic cases unless paired with the C282Y mutation.In the general population, 1 in 200 to 400 has two copies of the C282Y genetic mutation, thecombination of mutations which is most likely to cause symptoms of HFE-HHC.
Page 117 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Please bear in mind that most people who have these genetic mutations do not develop the disease.
The disease is less common among Hispanics, African Americans, Asians, and Native Americans.Roughly 13% of Hispanics, 8.5% of Asians, and 6% of African Americans is a carrier for the mildmutation, H63D. An additional 3% of Hispanics, 2.3% of African Americans are carriers of thepotentially disease-causing C282Y mutation.
How is HFE-associated hereditary hemochromatosisTreated?Ideally HFE-HHC is treated before the organs of the body are damaged. However, not everyone whohas the mutations that cause HFE-HHC develops symptoms or requires treatment. A simple blood testof iron levels in the blood—physicians specifically look at serum ferritin concentration and transferrin-iron saturation levels—can determine whether the body is absorbing too much. When iron reaches acertain threshold, treatment is recommended. If iron levels have not reached that threshold, notreatment is necessary. Blood tests must be repeated periodically to check these iron levels.
If a person has a high level of iron, treatment involves removing a certain quantity of blood at regularintervals. This is known as phlebotomy. Typically phlebotomy is performed frequently—perhapsweekly or twice weekly—until certain iron levels are reached, and then performed lessfrequently—often 2 to 4 times a year—on an indefinite basis. This treatment is simple, inexpensive,and safe.
If a person is already suffering from symptoms of HFE-HHC, treatment can lessen or relieve some ofthe symptoms. Cirrhosis is unlikely to improve with treatment, although treatment may slow itsprogression. If liver disease has reached severe levels, liver transplantation may be an option. Thosewho have any amount of liver damage are advised to avoid alcohol.
All people with symptoms of HFE-HHC are advised to eat only moderate amounts of iron-rich foods,avoid taking iron supplements or excess vitamin C, and refrain from eating uncooked seafood, as theyare highly susceptible to a particular kind of bacterial infection.
What is the Prognosis for Someone With HFE-associatedhereditary hemochromatosis?The prognosis for a person with the genetic mutations that cause HFE-HHC is generally good, as themajority of people in that situation do not develop symptoms of the disease. Most will not havedangerously elevated levels of iron in their blood, and therefore will not have any iron-overloadproblems.
For those that do have dangerously high iron levels in their blood, beginning treatment beforesymptoms appear is a critical part of ensuring a long, healthy life. Nearly all symptoms of the diseasecan be prevented with early and ongoing treatment. If a person with HFE-HHC is treated before he or
Page 118 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

she develops cirrhosis of the liver, he or she can expect a normal lifespan. Among people who alreadyhave cirrhosis associated with HFE-HHC, 72% will survive at least 5 more years and 62% will surviveat least 10 more years. People who already have cirrhosis are at an increased risk for developing a typeof liver cancer.
Page 119 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Homocystinuria Caused by CystathionineBeta-Synthase DeficiencyIncluding B6-Responsive Form and B6-Non-Responsive Form
Gene: CBS
Variant Accuracy PrecisionI278T 1.000 1.000G307S 1.000 1.000
Detection Rate Population36.60% African American38.00% Ashkenazi Jewish36.60% Eastern Asia38.00% Finland38.00% French Canadian or Cajun36.60% Hispanic36.60% Middle East36.60% Native American38.00% Northwestern Europe36.60% Oceania36.60% South Asia36.60% Southeast Asia38.00% Southern Europe
What is homocystinuria caused by cystathionine beta-synthase deficiency?Homocystinuria caused by cystathionine beta-synthase deficiency, or CBS deficiency, is an inheritedmetabolic disease in which affected people cannot process certain amino acids, the building blocks ofproteins, due to the lack of a particular enzyme. The disease causes an array of symptoms includingeye problems, skeletal abnormalities, an increased risk for dangerous blood clots, and in some people itcauses developmental delay or mental disability.
Not all people with the disease exhibit all of the symptoms described below, and the severity of thesymptoms varies widely from person to person. In some cases, people will reach adulthood beforedeveloping any symptoms.
There are two forms of the disease: a type that responds well to treatment with vitamin B6 and a typethat does not. People with B6-responsive CBS deficiency sometimes, but not always, have a lesssevere course of the disease than those with the B6-non-responsive form.
Page 120 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The majority of people with CBS deficiency are nearsighted as infants. Those who go untreated willexperience a dislocation of the lenses in their eyes (ectopia lentis) before the age of 8.
CBS deficiency can cause abnormal blood clots called thromboembolisms. If these blood clots form inor travel to the heart, brain, or other vital organs, they can be fatal. Thromboembolisms are frequentlythe cause of death in people with the disease.
People with the disease tend to be tall and thin with long arms and legs and an angular appearance.They are more likely than average to develop fragile, porous bones, a condition known as osteoporosis,at an early age. Roughly half of people with CBS deficiency will show signs of osteoporosis by theirteenage years. They are also prone to scoliosis, a curvature of the spine.
The IQs of some people with CBS deficiency have been recorded as high as 138, which is well abovethe general population average of 100 and would be considered “gifted.” The average IQ score for aperson with the disease, however, ranges between 57 and 79, depending on the genetic mutations he orshe inherited. These IQs are generally considered borderline to mild mental disability. IQs for peoplewith the disease have been recorded as low as 10, which indicates profound disability.
The disease can cause neurological or mental problems such as seizures, anxiety, depression,obsessive-compulsive behavior, and psychosis. Roughly half of people with CBS deficiency havesome kind of clinically significant psychiatric illness.
The disease can also cause abnormal muscle twitching, inflammation of the pancreas, the loss of skincolor, and liver problems.
How Common is homocystinuria caused by cystathioninebeta-synthase deficiency?The exact prevalence of homocystinuria caused by CBS deficiency is unknown. Studies have reportedthat 1 in every 250,000 people in the general population is affected by the disease, but this figure iswidely believed to be too low. Several studies estimate the populations in particular countries andregions as follows: Ireland (1 in 65,000), Germany (1 in 17,800), Norway (1 in 6,400), and Qatar (1 in3,000).
How is homocystinuria caused by cystathionine beta-synthase deficiency Treated?A main goal of treatment for a person with homocystinuria caused by CBS deficiency is to control theconcentration of homocysteine in the blood, thereby preventing symptoms of the disease. Medicalprofessionals can help design a particular diet which includes vitamins and supplements such asvitamin B6, vitamin B12, folate, and betaine, and avoids certain types of food, notably those with theamino acid methionine. It is important that this treatment begin in infancy so as to limit any damage tothe child’s body and intellect.
Page 121 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

In order to lower the risk of blood clots, people with the disease should avoid unnecessary surgery andwomen should not take oral contraceptives. Women with the disease who become pregnant shouldwork with their doctor to avoid pregnancy-related clotting problems.
Typically eye surgery is performed to correct the lens of the eye in people who have ectopia lentis.
What is the Prognosis for Someone With homocystinuriacaused by cystathionine beta-synthase deficiency?Homocystinuria with CBS deficiency is thought to shorten the lives of people affected by the disease,however there is no known average lifespan. About 1 in 4 people with the disease will die before age30 from a blood clot. The outlook is much better when the diagnosis is made early in life and theperson's diet can be carefully planned.
Page 122 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hurler SyndromeGene: IDUA
Variant Accuracy PrecisionA327P 1.000 1.000W402X 1.000 1.000
Detection Rate Population48.80% African American40.50% Ashkenazi Jewish48.80% Eastern Asia40.50% Finland40.50% French Canadian or Cajun48.80% Hispanic48.80% Middle East48.80% Native American40.50% Northwestern Europe48.80% Oceania48.80% South Asia48.80% Southeast Asia40.50% Southern Europe
What is Hurler syndrome?Hurler syndrome, also called mucopolysaccharidosis type I, is an inherited disease in which the bodylacks an enzyme called alpha-L-iduronidase. Without this enzyme, the body cannot properly breakdown long chains of sugar molecules called glycosaminoglycans. As a result, these moleculesaccumulate in the body, causing numerous health problems. Children with Hurler syndrome typicallydie before the age of 10, but may live longer with treatment.
Children with the disease appear normal at birth, but around the age of 9 months they typically begindeveloping some or all of the following symptoms:
• Appearance: Coarse facial features (broad mouth, square jaw), short neck, large head, small stature• Brain: Progressive and profound mental disability, tendency toward a dangerous accumulation of
fluid around the brain• Heart: Heart disease including valve problems and narrowed arteries• Eyes: Cloudy corneas leading to limited vision, glaucoma, and blindness• Bones: Spinal abnormalities, back pain, joint disease leading to restricted movement, claw hand,
carpal tunnel syndrome, misshapen bones• Ears: Moderate to severe hearing loss• Skin: Darkened areas
Page 123 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

• Digestive System: Enlarged liver and spleen, diarrhea and constipation• Lungs and Breathing: Progressive lung disease, frequent infection, chronic runny nose, airway
blockages, sleep apnea
A milder form of mucopolysaccharidosis type I is known as Scheie syndrome, and an intermediateform is known as Hurler-Scheie syndrome.
How Common is Hurler syndrome?Approximately 1 in 100,000 people have Hurler syndrome. It has been found in people of allethnicities.
How is Hurler syndrome Treated?Depending on the severity of Hurler syndrome and the age of the child, one of several treatments mayprevent or ameliorate some symptoms of the disease.
Bone marrow transplants can be effective in relieving physical aspects of Hurler syndrome, although itdoes not seem to help the bone or eye symptoms. Children who receive bone marrow transplantsearly—before the age of 2—tend to have better mental development, although they still have learningproblems and progressive mental decline. Outcomes of the procedure do vary, but a bone marrowtransplant can prolong the lifespan of a person with Hurler syndrome, even though it will still besignificantly shortened. Note that the procedure itself carries a high risk of fatality.
Umbilical cord blood is a more recent treatment for Hurler syndrome, allowing for an unrelated donorand eliminating the need for total body radiation, as is the norm with a bone marrow transplant. Thistreatment can prolong the lifespan of an affected child, but also does not help the bone and eye issues.A cord blood transplant can help prevent a certain measure of mental decline if it is performed beforesignificant damage is done to the intellect, often before the age of 18 months. Like bone marrowtransplants, the procedure itself carries a high risk of fatality and can result in a variety of outcomes.
Enzyme replacement therapy using recombinant human alpha-L-iduronidase (brand name:Aldurazyme) has also been shown to benefit people with Hurler syndrome, relieving many of thephysical symptoms. Enzyme replacement may be used in tandem with the above surgical options. Thistreatment is relatively new and further study is needed to determine its long-term success.
Other symptoms of the disease can be addressed as they arise. Examples of these treatments includespecial education for developmental delays, heart valve replacement, shunting to remove excess fluidand relieve pressure from around the brain, sunglasses or hats to promote better vision, and physicaltherapy to aid in movement.
Page 124 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With Hurlersyndrome?The prognosis for people with Hurler syndrome is generally poor. They need special education andassistance to perform ordinary daily functions, and are often wheelchair-bound. Death usually occurswithin the first 10 years of life, although early treatment such as a bone marrow transplant can extendthe lifespan. Heart and breathing problems are often the cause of death among children with thedisease.
Page 125 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hyperornithinemia-Hyperammonemia-Homocitrullinuria SyndromeGene: SLC25A15
Variant Accuracy PrecisionF188del 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland
95.00% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is hyperornithinemia-hyperammonemia-homocitrullinuria syndrome?Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH) syndrome is a rare inherited metabolicdisorder in which the body cannot properly process ammonia. Normally, the body converts ammoniainto urea, which can be excreted through urine. In people with HHH syndrome, ammonia and severalamino acids (ornithine and citrulline) build up in the body. High levels of ammonia can be damaging tothe brain and nervous system, causing movement problems and learning disabilities or loweredintelligence.
The exact symptoms and the age at which they begin vary widely among people with the disease. Insome cases, the disease can be fatal in infancy. In other cases, people with HHH syndrome have beenrelatively symptom-free until adulthood, when various neurological problems begin. If a low-proteindiet is begun early in life, many of the symptoms below can be avoided.
Infants with symptoms of HHH syndrome often vomit after consuming high-protein formula. Theymay also be lethargic and refuse to eat. Infants who are breast fed often do not show any symptomsuntil they begin to consume foods containing protein.
Page 126 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Children with HHH syndrome are prone to spastic movements in their hands and feet, poor muscletone, and seizures. Their growth and speech development may be delayed, along with other intellectualdevelopment milestones. They may have episodes of mental confusion, learning disabilities, and/or abelow-average IQ. These children often have poor performance and/or behavior problems in school.
When symptoms first appear in adults, they tend to arise as learning disabilities and episodes of blurredvision, confusion, dizziness, and/or spastic movement.
People with HHH syndrome tend to instinctively avoid high-protein foods such as meat or dairyproducts.
HHH syndrome belongs to a group of diseases known as urea cycle disorders.
How Common is hyperornithinemia-hyperammonemia-homocitrullinuria syndrome?HHH syndrome is extremely rare, with only about 50 cases known to physicians. Most of these caseshave occurred among French-Canadians in the province of Quebec, Canada.
How is hyperornithinemia-hyperammonemia-homocitrullinuria syndrome Treated?A strict low-protein diet begun early in life can prevent many or all symptoms of the disease. Peoplewith HHH syndrome should work with a nutritionist to design a diet that eliminates harmful proteinwhile still allowing for needed nutrients. Some people with the disease do not respond to changes indiet and still experience symptoms of HHH syndrome.
To reduce ammonia in the body, some people benefit from taking supplements of the amino acidsornithine or citrulline. Sodium benzoate and sodium phenylacetate may also be recommended by adoctor.
What is the Prognosis for Someone Withhyperornithinemia-hyperammonemia-homocitrullinuriasyndrome?The prognosis for people with HHH syndrome varies widely. If the disease is detected and treatedearly, it is possible to live a normal life with few, if any, symptoms of the disease. In some people, thedisease causes varying degrees of neurological problems, which can be progressive. If untreated, manypeople with the disease will have shortened lifespans.
It is possible for women with the disease to give birth to healthy babies.
Page 127 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Hypophosphatasia, Autosomal RecessiveGene: ALPL
Variant Accuracy Precision1559delT 1.000 1.000F310L 1.000 1.000D361V 1.000 1.000E174K 1.000 1.000G317D 1.000 1.000
Detection Rate Population<10% African American
22.23% Ashkenazi Jewish53.50% Eastern Asia22.23% Finland22.23% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
22.23% Northwestern Europe<10% Oceania<10% South Asia
53.50% Southeast Asia22.23% Southern Europe
What is hypophosphatasia, autosomal recessive?Hypophosphatasia is an inherited disorder that disrupts a process called mineralization, in which thebody deposits minerals like calcium and phosphorus into teeth and bones. Proper mineralization isnecessary to make bones strong and rigid, and make teeth strong enough to withstand years ofchewing.
Hypophosphatasia can have two types of inheritance patterns: autosomal recessive (symptoms are seenwhen both disease genes have mutations) or autosomal dominant (symptoms occur when only one oftwo disease genes has a mutation). With autosomal recessive hypophosphatasia, symptoms can varygreatly depending upon which mutations a person carries. Some forms of the disease are severe whileother forms are extremely mild.
The most severe form of hypophosphatasia appears before birth or in early childhood. In many cases,infants are stillborn because their skeletons fail to form. Other affected infants are born with shortlimbs, soft skull bones, and an abnormally shaped chest caused by soft, weak ribs. Approximately halfthe infants born with the condition die of respiratory failure in the first few weeks of life. Those who
Page 128 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

survive may have life-threatening complications such as breathing problems, seizures, or high bloodcalcium levels leading to kidney damage.
In a less severe form, children show the first signs of the condition by losing their baby teeth before theage of five. As they grow, they may be below average in height, with bowed legs or knock knees, largewrist and ankle joints, and an abnormally shaped skull. They are more prone to broken bones, bonepain, and arthritis. They may have trouble learning to walk or may develop a waddling gait. Their teethmay crack or decay more easily than normal.
The mildest form of the disorder is called odontohypophosphatasia. It only affects the teeth. Peoplewith this form of the condition have abnormal tooth development and lose their teeth early, but do nothave skeletal abnormalities.
Occasionally, people with hypophosphatasia do not develop any symptoms until middle age. The mostcommon symptoms are early tooth loss and frequent, slow-healing stress fractures in the feet. Theymay also develop arthritis.
How Common is hypophosphatasia, autosomalrecessive?Hypophosphatasia affects approximately 1 in 100,000 people and is most common in Caucasians. Thedisease is particularly common in a particular Mennonite population in Ontario, Canada, where itaffects 1 in 2,500 people.
How is hypophosphatasia, autosomal recessive Treated?Infants with the most severe form of the condition usually require mechanical help to breathe and mayneed surgery to release pressure within the skull. Vitamin B6 may relieve seizures.
Children and adults with hypophosphatasia should see a dentist every year, beginning at the age of one,to preserve teeth as long as possible. Adults will eventually need false teeth.
Aspirin, ibuprofen, and other pain relievers help with bone pain and arthritis. Although preventingbone fractures is difficult, orthotics may help with common fractures in the feet.
People with the condition should not take bisphosphonates, which are drugs commonly prescribed totreat other bone loss conditions such as osteoporosis. They should also avoid excess vitamin D, whichcan make calcium build up in the blood.
Page 129 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone Withhypophosphatasia, autosomal recessive?Approximately 50% of infants born with the severe form of the condition will die of respiratory failurein infancy. Exact lifespan for the rest is not known. People with the milder forms of the condition havenormal lifespans.
Page 130 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Inclusion Body Myopathy 2Gene: GNE
Variant Accuracy PrecisionM712T 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is inclusion body myopathy 2?Inclusion body myopathy 2 (IBM2) is an inherited disease that causes a progressive weakening of thelegs and arms, typically beginning in the late teens or early 20s and almost always before the age of 40.Typically people with the disease lose the ability to walk 20 years after symptoms appear.
The muscles of the lower leg are typically affected first. As these muscles slowly weaken, walkingbecomes more difficult and the person’s gait changes. The weakness will spread to the thighs, handmuscles, and certain muscles of the shoulder and neck. A small number of people will also haveweakness in the facial muscles. Often the large thigh muscles (quadriceps) are unaffected until late inthe course of the disease.
For reasons not well understood, a small number of people who have the genetic mutations that causeIBM2 do not have symptoms of the disease.
How Common is inclusion body myopathy 2?IBM2 is most common among Middle Eastern Jews, particularly of Iranian descent. The disease hasalso been found in small numbers of non-Jews, both within and outside of the Middle East. Roughly220 individuals with IBM2 have been reported in medical literature, making the disease very rare inthe general population.
Page 131 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Studies estimate that among Iranian Jewish communities in Israel and Los Angeles, 1 in 15 people arecarriers of mutations that cause IBM2. These studies also estimate that 1 in every 500 to 1000 IranianJews in these communities are affected by IBM2.
How is inclusion body myopathy 2 Treated?There is no cure or treatment for IBM2 that can reverse or delay the progression of muscle weakness.Neurologists, rehabilitation specialists, and physical and occupational therapists can aid in relievingsymptoms as they appear.
What is the Prognosis for Someone With inclusion bodymyopathy 2?The disease often does not cause noticeable symptoms until the late teens or early 20s when muscleweakness begins. Movement of the arms and legs will become progressively impaired and typicallypeople with IBM2 are wheelchair-bound 20 years after symptoms begin. The disease's effect onlifespan is not well-studied.
Page 132 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Infantile Refsum DiseaseGene: PEX1
Variant Accuracy PrecisionG843D 1.000 1.000
Detection Rate Population43.10% African American43.10% Ashkenazi Jewish43.10% Eastern Asia43.10% Finland43.10% French Canadian or Cajun43.10% Hispanic43.10% Middle East43.10% Native American43.10% Northwestern Europe43.10% Oceania43.10% South Asia43.10% Southeast Asia43.10% Southern Europe
What is infantile Refsum disease?Infantile refsum disorder (IRD) causes developmental delays leading to mental disability, hearing andvision impairment, liver problems, skeletal abnormalities, and episodes of spontaneous bleeding, oftenaround the brain. The disease is often fatal in childhood.
Symptoms of IRD vary from person to person and may not include all of the symptoms listed above. Inthe mildest cases of IRD, the child has mild learning disabilities and mild vision and hearing loss.Typically these symptoms begin in infancy and grow more severe over time.
Many children with the disease learn to walk, while others lack the muscle tone needed for suchmovement. Similarly many children with the disease learn to talk, though some do not.
IRD's symptoms are caused by an inability to properly break down phytanic acid, a substance commonin foods such as dairy products, beef, lamb, and tuna. Phytanic acid then accumulates to high levels inthe body, causing the characteristic symptoms.
IRD is the mildest subtype of Zellweger syndrome spectrum and belongs to a group of diseases knownas peroxisome biogenesis disorders. These are part of an even larger group of diseases known asleukodystrophies.
Page 133 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is infantile Refsum disease?IRD is a subtype of Zellweger syndrome spectrum (ZSS). Overall, ZSS affects an estimated 1 in50,000 to 100,000 infants. However, it may be less common in Asia, where one study estimated that 1in 500,000 Japanese infants are affected. IRD is thought to account for about 40% of ZSS cases.
How is infantile Refsum disease Treated?There is no cure for IRD. Physicians typically address symptoms of the disease as they appear.
In order to minimize damage to the body, children with IRD should avoid eating foods containingphytanic acid. These include (but are not limited to) dairy products, lamb and beef, and fish such astuna, cod, and haddock.
Children with IRD may benefit from hearing aids, eye glasses, and/or surgery to remove cataracts. Inthose who reach school age, special education is likely necessary.
Some children with IRD may benefit from treatments that remove blood, filter out phytanic acid, andreturn the blood to the body.
What is the Prognosis for Someone With infantile Refsumdisease?In people with IRD, lifespan will vary depending on the severity of the symptoms they experience.Many people with the disease die in childhood, though some live into their teens and twenties andpossibly beyond. One study showed that among children with milder forms of Zellweger syndromespectrum (this includes IRD and another condition known as neonatal adrenoleukodystrophy) whosesymptoms did not worsen over time, 77% who reached the age of one year survived until they wereschool age.
Children with IRD all have some degree of learning disability or mental disability as well as somedegree of hearing and sight impariment.
Page 134 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Isovaleric AcidemiaGene: IVD
Variant Accuracy PrecisionA311V 1.000 1.000
Detection Rate Population47.00% African American47.00% Ashkenazi Jewish47.00% Eastern Asia47.00% Finland47.00% French Canadian or Cajun47.00% Hispanic47.00% Middle East47.00% Native American47.00% Northwestern Europe47.00% Oceania47.00% South Asia47.00% Southeast Asia47.00% Southern Europe
What is isovaleric acidemia?Isovaleric acidemia (IVA) is an inherited disorder in which the body is unable to properly processproteins, leading to a toxic buildup of isovaleric acid in the blood.
Some people with the genetic mutations that cause IVA do not show symptoms of the disease. Thereason for this lack of symptoms is not entirely understood.
As the body digests proteins, it breaks them into smaller parts called amino acids. People with IVAlack a properly-working enzyme necessary to break down a common amino acid called leucine. As aresult, an organic acid called isovaleric acid reaches toxic levels in the blood and can cause damage tothe brain and nervous system. If left untreated, IVA can lead to seizures, coma, and death. Treatmentwith an appropriate low-protein diet, however, can lead to fairly normal growth, development, andlifespan.
There are two forms of IVA, one which appears shortly after birth and is rapidly life-threatening, andanother which may appear later in childhood as episodes of illness.
The form of IVA seen in newborns appears within two weeks of birth. Initial symptoms include lack ofenergy, poor appetite, vomiting, and difficulty staying warm. The buildup of isovaleric acid often givesthem an odor of sweaty feet. If untreated, these infants will progress to a more serious metabolic crisis,suffering seizures, coma, swelling or bleeding of the brain, and even death.
Page 135 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The childhood form of IVA often appears around the child's first birthday and its symptoms—nearlyidentical to the infantile form of the disease—may come and go over time, flaring up particularlyduring times of illness, high protein consumption, or long periods without food. Between periods ofcrisis, the child can be healthy, however overall these children may show poor growth, muscleweakness, or learning problems. Some infants who have the more severe, early-onset form of thedisease can progress to the more episodic form later in life.
IVA is part of a group of diseases known as organic acid disorders.
How Common is isovaleric acidemia?IVA affects at least 1 in 250,000 Americans.
How is isovaleric acidemia Treated?People with IVA must eat a diet low in proteins, minimizing foods such as dairy products, meat, fish,eggs, legumes, and nuts. The body does need protein, however, and a nutritionist or other medicalprofessional can help devise an appropriate diet. This may include foods made especially for peoplewith organic acid disorders. Supplements of carnitine and/or glycine may be prescribed. Thesesupplements bind with isovaleric acid and turn it into a less harmful compound.
People with IVA should have close contact with a physician during times of illness. At these times, thebody may break down its own protein, leading to a buildup of isovaleric acid. Typically people withIVA need to eat more carbohydrates and drink more fluids during times of illness, even if they are nothungry or thirsty.
Any time a child with IVA experiences a metabolic crisis, he or she needs prompt treatment, whichmay include a hospital visit.
What is the Prognosis for Someone With isovalericacidemia?If the disease is recognized promptly and treated diligently, children with IVA can live near-normallives. It is possible, however, that they will have episodes of metabolic crisis, although these episodestend to decrease with age. If these episodes are not treated, irreversible learning problems or mentaldisability can occur.
Those who do not develop any symptoms of the disease can be expected to live a normal lifespan.
Page 136 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Krabbe DiseaseIncluding Infantile Form and Late-Onset Form
Gene: GALC
Variant Accuracy PrecisionEx11-17del 1.000 1.000G270D 1.000 1.000R168C 1.000 1.000
Detection Rate Population<10% African American
46.50% Ashkenazi Jewish<10% Eastern Asia
46.50% Finland46.50% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
46.50% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
46.50% Southern Europe
What is Krabbe disease?Krabbe disease, also known as globoid cell leukodystrophy, is an inherited degenerative disease of thenervous system. Leukodystrophies are a group of diseases which affect the myelin sheath, a fattycovering that insulates and protects nerve cells. People with Krabbe disease lack an enzyme calledgalactocerebrosidase, and the result is a build-up of toxic substances in cells that produce the myelinsheath. Without this protective covering, brain cells die and nerves in the body cannot functionproperly.
There are two forms of the disease: infantile and late-onset.
Infantile Form
The infantile form, which affects 85 to 90% of people with Krabbe disease, appears in the first fewmonths of life and causes irritability, muscle weakness, unexplained fever, deafness, blindness,seizures, and slowed mental and physical development. Usually death occurs by the age of two, oftendue to respiratory failure.
Page 137 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Late-onset Form
The late onset form of Krabbe disease, which affects 10 to 15% of people with the disease, can appearat any time between the ages of six months and fifty years. These individuals slowly develop visionloss, difficulty walking, rigid muscles, and mental impairment. Symptoms among people with lateonset Krabbe disease are highly variable. The disease is often fatal 2 to 7 years after symptoms begin.
How Common is Krabbe disease?About 1 in 100,000 people in the United States and Europe have Krabbe disease, and 1 in 150 arethought to be carriers. Several Druze and Muslim communities in and around Israel have anabnormally high incidence of Krabbe disease. There, as many as 1 in 6 adults may be carriers of thedisease.
How is Krabbe disease Treated?Treatment for Krabbe disease will depend on which form of the disease a person has. Treatmentoptions for both forms are listed below.
Infantile Form
For infants with this form of Krabbe disease who have not yet started showing symptoms, treatmentwith umbilical cord blood stem cells has shown promise in enabling normal or near normal lives. Thisprocedure can take place within weeks of birth. In many cases neural deterioration is slowed followingthe procedure and symptoms seem less severe.
Bone marrow stem cells may be used in place of umbilical cord blood stem cells, however cord bloodstem cells are less particular and do not require the donor to be a perfect match. With cord blood stemcells, there is also less risk of immune system complications.
Infants who have already started showing symptoms of the disease do not seem to benefit from thistreatment. For them and others not suitable for the procedure, the only treatment is to addresssymptoms as they arise.
Late-onset Form
Some people with late onset Krabbe disease have benefited from treatment with umbilical cord stemcells, although this treatment has been most successful in pre-symptomatic patients with the infantileform of the disease. In cases where the treatment has been successful, neural deterioration is slowedand symptoms are less severe.
Page 138 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Bone marrow stem cells may be used in place of umbilical cord blood stem cells, however cord bloodstem cells are less particular and do not require the donor to be a perfect match. With cord blood stemcells, there is also less risk of immune system complications.
For those not suitable for the procedure, the only treatment is to address symptoms as they arise.
What is the Prognosis for Someone With Krabbedisease?The infantile form of Krabbe disease is usually fatal before the age of two. Those infants who receivecord blood stem cells before the appearance of symptoms have longer lifespans.
Those with late-onset Krabbe disease typically live between 2 and 7 years after the onset of symptoms.The exact symptoms and rate of neurological deterioration varies greatly from person to person, evenamong those in the same family who have the same genetic mutations.
Page 139 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Leigh Syndrome, French-Canadian TypeGene: LRPPRC
Variant Accuracy PrecisionA354V 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland
97.70% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Leigh syndrome, French-Canadian type?Leigh syndrome, French-Canadian type, is an inherited disease that causes degeneration of the brain aswell as metabolic abnormalities. It is caused by a deficiency in the cytochrome oxidase (COX) enzymecomplex and is usually fatal by the age of 5 or 6.
The disease primarily affects people from the Saguenay Lac-Saint-Jean region of Quebec, Canada andis rare elsewhere.
The French-Canadian form of Leigh syndrome causes developmental delay, poor muscle tone, crossedeyes, characteristic facial features, and a tendency toward life-threatening metabolic crisis and coma. Itcauses characteristic lesions on the brain and spinal cord. The disease shares symptoms with otherforms of Leigh syndrome, which cause progressive degeneration in brain tissues.
Metabolic crises can cause low blood sugar and an extreme lack of energy and can very quickly turnfatal. Often illness can precipitate a metabolic crisis.
How Common is Leigh syndrome, French-Canadian type?Among French-Canadians in the Saguenay Lac-Saint-Jean region of Quebec, Canada, 1 in every 22 to23 people are carriers of this form of Leigh syndrome. As a result, roughly 1 in 2,000 infants in thisregion is affected by the disease.
Page 140 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Outside this region of Quebec, the disease is rare.
How is Leigh syndrome, French-Canadian type Treated?There are no successful treatments for Leigh syndrome, French-Canadian type.
What is the Prognosis for Someone With Leighsyndrome, French-Canadian type?The prognosis for a person with Leigh syndrome, French-Canadian type is poor. The median lifeexpectancy is 5 to 6 years.
Page 141 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Limb-Girdle Muscular Dystrophy Type 2EGene: SGCB
Variant Accuracy PrecisionS114F 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is limb-girdle muscular dystrophy type 2E?Limb-girdle muscular dystrophy type 2E (LGMD2E), also known as beta-sarcoglycanopathy, is aninherited disease in which muscles of the hip, abdomen, and shoulder progressively weaken.
Symptoms of the disease vary greatly from person to person, even among people in the same family.LGMD2E is often associated with severe symptoms that can be fatal by the late teens, although somepeople with the disease have a mild course or are nearly asymptomatic.
People with LGMD2E commonly develop symptoms before the age of 10, although in some cases,symptoms do not appear until later. Typically the later in life symptoms appear, the less rapidly theywill progress. In people with LGMD2E, the muscles of the hip, shoulder, and abdomen progressivelyweaken, often to a point where a wheelchair becomes necessary. This typically happens between theages of 10 and 15.
In people with LGMD2E, muscles attached to the scapula, a bone that connects the arm with the collarbone, do not work properly, causing a wing-like shape to the upper back. The calf muscles are oftenenlarged as well, a symptom known as calf hypertrophy.
A minority of people with LGMD2E — about 20% — experience a weakening of the heart muscles.Involvement of the heart muscles is less common in type 2E than in other forms of limb girdlemuscular dystrophy.
Page 142 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

LGMD2E does not affect intelligence or mental function.
The cause of LGMD2E is a deficiency in a protein called beta-sarcoglycan. The function of thisprotein is not well understood. The degree of beta-sarcoglycan deficiency may correlate with theseverity of disease symptoms.
How Common is limb-girdle muscular dystrophy type 2E?There are numerous types of limb-girdle muscular dystrophy, and combined worldwide, they occur in5 to 70 of every 1,000,000 people. LGMD2E is rare, but its exact prevalence is unknown.
How is limb-girdle muscular dystrophy type 2E Treated?There is no cure for LGMD2E and few effective treatments. Physical therapy is often recommended toretain muscle strength and mobility for as long as possible. Stretching, mechanical aids, or surgery mayaid in this goal. Those who develop heart problems should consult with a cardiologist for symptomatictreatments. As muscles deteriorate, a ventilator may be required to aid breathing.
What is the Prognosis for Someone With limb-girdlemuscular dystrophy type 2E?The outlook for a person with LGMD2E is varied. Generally speaking, the earlier symptoms begin, thefaster they progress. Some people with the disease experience only mild symptoms, and may havenear-normal strength. In these cases, the disease may not affect lifespan. In general, many people withthe disease live into their 30s. People with more severe symptoms can become wheelchair boundbetween the ages of 10 and 15 and die in their late teens, often as a result of respiratory failure.
Page 143 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Long Chain 3-Hydroxyacyl-CoADehydrogenase DeficiencyGene: HADHA
Variant Accuracy PrecisionQ342X 1.000 1.000E474Q 1.000 1.000
Detection Rate Population83.33% African American82.33% Ashkenazi Jewish83.33% Eastern Asia
>99% Finland82.33% French Canadian or Cajun83.33% Hispanic83.33% Middle East83.33% Native American82.33% Northwestern Europe83.33% Oceania83.33% South Asia83.33% Southeast Asia82.33% Southern Europe
What is long chain 3-hydroxyacyl-CoA dehydrogenasedeficiency?Long chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency is an inherited condition inwhich the body lacks sufficient amounts of an enzyme needed to turn certain fatty acids from food andbody fat into energy. This process, called fatty acid oxidation, normally breaks down fatty acidsstepwise until they can be turned into usable energy. LCHAD deficiency interrupts this process,allowing fatty acids to build up in the body and damage various organs and body tissues.
The symptoms of LCHAD deficiency begin in infancy or early childhood when affected children maydisplay muscle weakness, lack of energy, low blood sugar, liver problems, and difficulty feeding. Theyare at high risk for life-threatening heart and breathing problems, comas, and seizures, as well assudden unexplained infant death.
Children with LCHAD deficiency tend to experience symptoms in bouts, followed by periods ofseeming normalcy. Repeated episodes, if not properly treated, can cause brain damage, learningdisabilities, or mental disability. Periods of fasting, illness, or strenuous exercise can instigate orexacerbate these episodes.
Page 144 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Later in childhood, people with LCHAD often develop muscle pain, a breakdown in muscle tissue, andproblems with the nerves in their arms and legs. The disease can also cause damage to the retina of theeye, causing progressive visual impairment over many years.
Pregnant women whose fetuses are affected by LCHAD deficiency are at risk for specific pregnancycomplications and should speak with their physician.
How Common is long chain 3-hydroxyacyl-CoAdehydrogenase deficiency?LCHAD deficiency is rare. Studies in Finland and the United States have shown that the E474Qmutation—which causes the majority of LCHAD deficiency cases—has a carrier rate of approximately1 in 150 to 200. Based on a carrier rate estimation of 1 in 150, 1 in 90,000 people would be affected bythe disease.
How is long chain 3-hydroxyacyl-CoA dehydrogenasedeficiency Treated?The main method of treatment for LCHAD deficiency is to monitor the diet very carefully. Aphysician or nutritionist will devise a course of treatment that normally involves frequent meals, oftenaround the clock. There may need to be an additional feeding schedule for times when the person issick. The diet is often low in fats and high in carbohydrates, which are easier for an affected person tobreak down. A physician may also prescribe medium chain triglyceride oil, L-carnitine, and/or othersupplements for additional energy. People with LCHAD deficiency may need to curtail their exerciseor exposure to extreme cold.
What is the Prognosis for Someone With long chain3-hydroxyacyl-CoA dehydrogenase deficiency?With careful monitoring of diet, people with the disease can live long, near-normal lives. Despitecareful treatment, they may experience periodic problems in body chemistry that can result in braindamage, learning disabilities, or mental disability as well as problems with the muscles, heart, liver,and/or vision. If untreated, LCHAD deficiency can be fatal.
Page 145 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Maple Syrup Urine Disease Type 1BGene: BCKDHB
Variant Accuracy PrecisionG278S 1.000 1.000E322X 1.000 1.000R183P 1.000 1.000
Detection Rate Population<10% African American
87.00% Ashkenazi Jewish<10% Eastern Asia
40.05% Finland40.05% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
40.05% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
40.05% Southern Europe
What is maple syrup urine disease type 1B?Maple syrup urine disease (MSUD) type 1B is an inherited metabolic disorder named for thecharacteristic maple syrup smell of the affected person’s urine. If carefully treated with a low-proteindiet, people with MSUD can live fairly normal lives.
MSUD is caused by the lack of an enzyme needed to break down three amino acids: leucine,isoleucine, and valine, which are collectively known as the branched-chain amino acids. These aminoacids are found in all foods containing protein. Without the needed enzyme, known as branched-chainketoacid dehydrogenase (BCKAD), these amino acids and their byproducts accumulate and causedamage to the body. MSUD type 1B is due to a defect in one of the four components of the BCKADenzyme.
Maple syrup urine disease can be classified into four general types: classic, intermediate, intermittent,and thiamine-responsive. Classic MSUD is the most severe type. People with other types exhibitmilder symptoms, but are prone to periods of crisis in which symptoms closely resemble classicMSUD. In all types of the disease, there is a risk of mental and physical disability.
Page 146 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Classic
The most common type, classic MSUD is characterized by little or no enzyme activity. Symptoms inpeople with classic MSUD will appear in the first week of life. Within 12 to 24 hours, or upon firstconsumption of protein, the infant’s urine will take on a maple syrup smell. (Mediterraneanpopulations unfamiliar with maple syrup describe the odor as similar to fenugreek.)
Within several days, the infant will show poor feeding, vomiting, and irritability, followed by lack ofenergy, weight loss, seizures, a tense arched posture, muscle tone which alternates between stiff andlimp, and swelling of the brain. If untreated, life-threatening coma or respiratory failure could occurwithin 7 to 10 days and most will die within several months.
Upon any lapse of treatment, classic MSUD can cause brain damage. People with the disease areparticularly prone to crisis during illness, infection, fasting, or after surgery.
Intermediate
People with intermediate MSUD have 3 to 8% of the normal amount of BCKAD enzyme activity. As aresult, their bodies can tolerate higher amounts of the amino acid leucine. When ill, however, thistolerance drops.
Intermediate MSUD is similar to but less severe than the classic form. During periods of crisis,however, symptoms and risks are nearly identical.
Intermittent
With intermittent MSUD, BCKAD enzyme activity is often between 8 and 15% of normal. Symptomsof the disease may not appear until the first or second year of life. Symptoms often appear duringillness, fasting, or periods of high protein consumption.
This form of the disease is rare, but in times of crisis its risks and symptoms are similar to the classicform.
Thiamine-Responsive
Thiamine-responsive MSUD is distinct in that people with this form of the disease will respond tolarge doses of thiamine. One study found that people with thiamine-responsive MSUD have 30 to 40%the normal activity of the BCKAD enzyme. Many people with this form of the disease can toleratesome protein in their diet.
In times of crisis, the risks and symptoms of thiamine-responsive MSUD are similar to the classicform. The ability to treat the disease with thiamine, however, makes it easier to control than the otherforms, whose treatment hinges largely on diet.
Page 147 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is maple syrup urine disease type 1B?Worldwide, MSUD type 1B is estimated to affect 1 in 185,000 infants. It is most common among theOld Order Mennonite population, where about 1 in 385 infants is affected by the disease. AmongMennonites of eastern Pennsylvania, the frequency has been reported as high as 1 in 176 infants. Thedisease is also more common among Ashkenazi Jews, with roughly 1 in 50,000 affected by MSUDtype 1B.
How is maple syrup urine disease type 1B Treated?MSUD type 1B is primarily controlled by diet, using foods low in protein. This often means severerestrictions on meat, fish, eggs, dairy foods, wholegrain flour, beans, and nuts. Often people withMSUD type 1B are given a special liquid formula that supplies nutrients without the amino acids theycannot digest. These dietary restrictions should begin immediately upon diagnosis and must continuefor the person’s entire life.
Amino acid levels in the blood should be monitored regularly by a physician. Blood test findings canhelp to calibrate the diet, and are particularly important during pregnancy for a mother with MSUD.Any swelling of the brain requires immediate medical attention. Illnesses should always prompt aconsultation with a physician, as these are vulnerable periods for a person with MSUD type 1B. He orshe may need a special “sick day diet” to avoid hospital stays.
Those with thiamine-responsive MSUD may be prescribed thiamine supplements.
What is the Prognosis for Someone With maple syrupurine disease type 1B?With early, careful, and lifelong treatment, people with MSUD type 1B can live healthy lives intoadulthood and show normal growth and mental development. It is particularly critical to recognize thedisease as soon as symptoms appear in order to avoid brain damage and mental disability. Despitecareful treatment, some people with the disease will experience periodic flare-ups, particularly duringtimes of illness. These may create learning problems or mental disability and can be life-threatening.
If untreated, MSUD can be fatal.
Page 148 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Maple Syrup Urine Disease Type 3Gene: DLD
Variant Accuracy Precision105insA 1.000 1.000G229C 1.000 1.000
Detection Rate Population<10% African American>99% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is maple syrup urine disease type 3?Maple syrup urine disease (MSUD) type 3 is a rare inherited disease that causes metabolicabnormalities, neurological damage, poor muscle tone, developmental delay, and movement problems.In some people, it is fatal in childhood. Infants with MSUD type 3 often appear normal until the age of8 weeks to 6 months when they develop severe lactic acidosis, a buildup of lactic acid in the body thatcauses vomiting, abdominal pain, and rapid breathing. If untreated, it can be fatal.
In addition to lactic acid buildup, a number of other substances accumulate in the bodies of people withMSUD type 3. These include blood pyruvate, alpha-ketoglutarate, branched-chain amino acids, alpha-hydroxyisovalerate, and alpha-hydroxyglutarate.
Infants and children with the disease show developmental delay and a progressive breakdown of theirnervous system. They often have poor muscle tone (hypotonia) and abnormal movements. Their urinetakes on a characteristic "maple syrup" smell for which the disease is named.
How Common is maple syrup urine disease type 3?MSUD type 3 is extremely rare. Fewer than 20 cases are known worldwide. The majority of knowncases come from families of Ashkenazi Jewish background.
Page 149 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is maple syrup urine disease type 3 Treated?There is no established treatment for MSUD type 3 due to the rarity of the disease. Combinations ofdiet, vitamins, and supplements have been tried without much success.
What is the Prognosis for Someone With maple syrupurine disease type 3?While the number of known cases does not allow for a well-established prognosis, it is thought thatmost people with MSUD type 3 will die during childhood.
Page 150 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
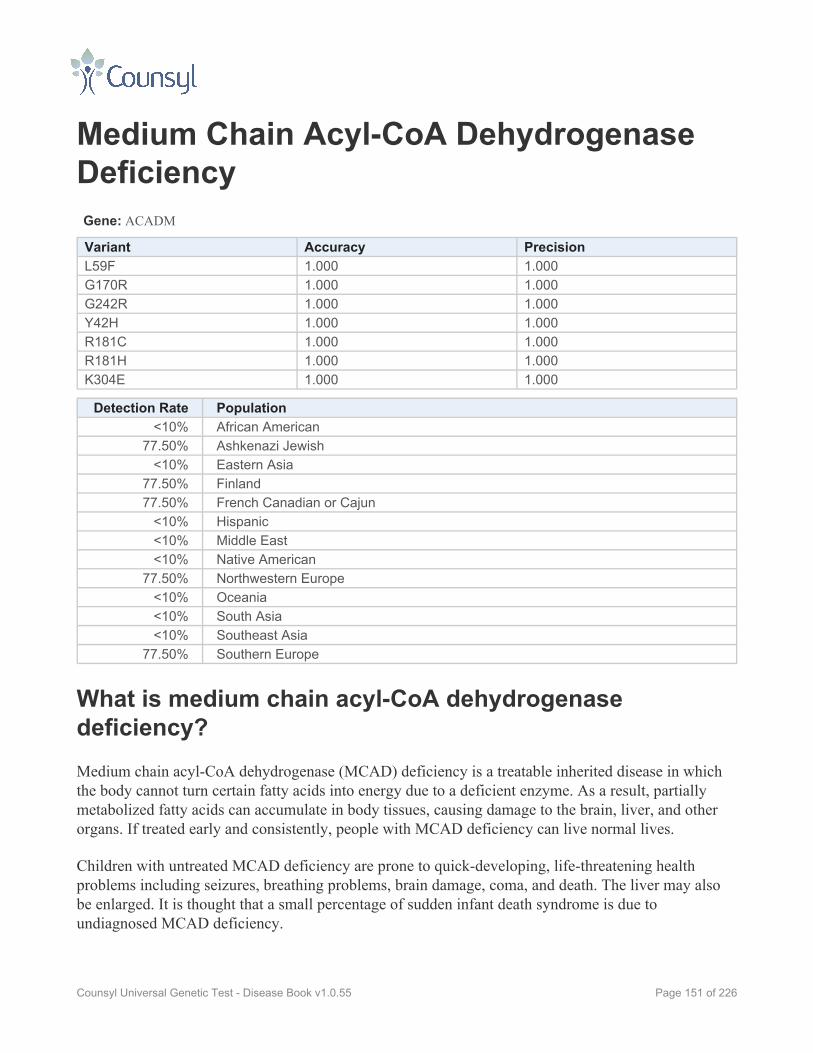
Medium Chain Acyl-CoA DehydrogenaseDeficiencyGene: ACADM
Variant Accuracy PrecisionL59F 1.000 1.000G170R 1.000 1.000G242R 1.000 1.000Y42H 1.000 1.000R181C 1.000 1.000R181H 1.000 1.000K304E 1.000 1.000
Detection Rate Population<10% African American
77.50% Ashkenazi Jewish<10% Eastern Asia
77.50% Finland77.50% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
77.50% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
77.50% Southern Europe
What is medium chain acyl-CoA dehydrogenasedeficiency?Medium chain acyl-CoA dehydrogenase (MCAD) deficiency is a treatable inherited disease in whichthe body cannot turn certain fatty acids into energy due to a deficient enzyme. As a result, partiallymetabolized fatty acids can accumulate in body tissues, causing damage to the brain, liver, and otherorgans. If treated early and consistently, people with MCAD deficiency can live normal lives.
Children with untreated MCAD deficiency are prone to quick-developing, life-threatening healthproblems including seizures, breathing problems, brain damage, coma, and death. The liver may alsobe enlarged. It is thought that a small percentage of sudden infant death syndrome is due toundiagnosed MCAD deficiency.
Page 151 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

The first symptoms of the disease usually appear in infancy or early childhood. These includevomiting, lack of energy, and low blood sugar. Rarely, these symptoms do not appear until adulthood.Often the episodes of metabolic crisis can be triggered by long periods without eating or by illness.
Women whose fetuses have MCAD deficiency are more prone to certain pregnancy complications andshould speak with their physician.
How Common is medium chain acyl-CoA dehydrogenasedeficiency?MCAD deficiency is most common in Caucasians from Northern Europe. In the United States, thedisease affects approximately 1 in 17,000 people. Affected Americans are often of Northern Europeanancestry. The disease is rare among Hispanics, African Americans, Asians, and Native Americans inthe United States.
Studies have found high rates of MCAD deficiency in Northern Germany (1 in 4,900) and SouthernGermany (1 in 8,500). One study found that Germans and Turks are equally affected.
How is medium chain acyl-CoA dehydrogenasedeficiency Treated?The key to treatment for people with MCAD deficiency is to avoid fasting, or long periods withouteating. Infants will need to be fed frequently with a special diet low in fat. Consuming cornstarch canprovide a sustained release of energy and allow for longer gaps between meals. Certain types of fatshould be avoided while high amounts of carbohydrates can be beneficial. If the person is unable toconsume food, intravenous glucose may be necessary. People with MCAD deficiency should speakwith their medical team to devise a specialized diet.
What is the Prognosis for Someone With medium chainacyl-CoA dehydrogenase deficiency?If a person affected by MCAD deficiency is diagnosed early and treated promptly, the prognosis isgood. He or she can lead a normal or near-normal life.
Because the metabolic crises caused by the disease can quickly progress from first symptom to death,it is possible for people who remain undiagnosed to die during their first episode.
Page 152 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
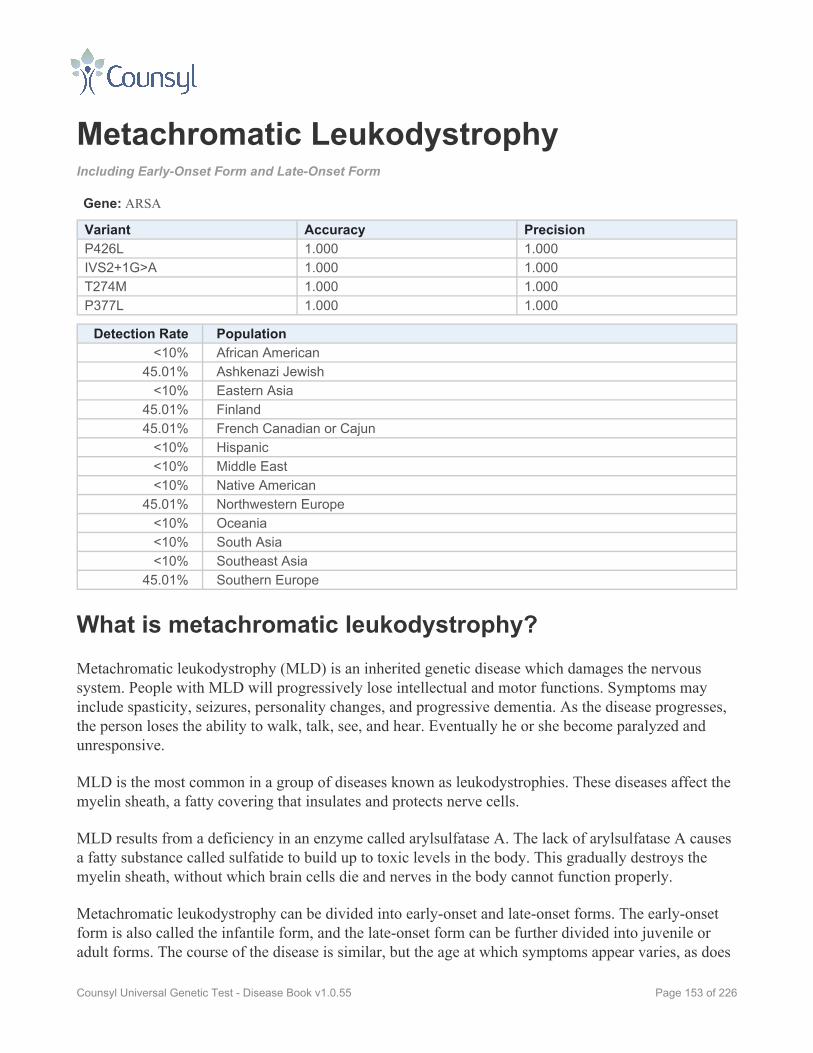
Metachromatic LeukodystrophyIncluding Early-Onset Form and Late-Onset Form
Gene: ARSA
Variant Accuracy PrecisionP426L 1.000 1.000IVS2+1G>A 1.000 1.000T274M 1.000 1.000P377L 1.000 1.000
Detection Rate Population<10% African American
45.01% Ashkenazi Jewish<10% Eastern Asia
45.01% Finland45.01% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
45.01% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
45.01% Southern Europe
What is metachromatic leukodystrophy?Metachromatic leukodystrophy (MLD) is an inherited genetic disease which damages the nervoussystem. People with MLD will progressively lose intellectual and motor functions. Symptoms mayinclude spasticity, seizures, personality changes, and progressive dementia. As the disease progresses,the person loses the ability to walk, talk, see, and hear. Eventually he or she become paralyzed andunresponsive.
MLD is the most common in a group of diseases known as leukodystrophies. These diseases affect themyelin sheath, a fatty covering that insulates and protects nerve cells.
MLD results from a deficiency in an enzyme called arylsulfatase A. The lack of arylsulfatase A causesa fatty substance called sulfatide to build up to toxic levels in the body. This gradually destroys themyelin sheath, without which brain cells die and nerves in the body cannot function properly.
Metachromatic leukodystrophy can be divided into early-onset and late-onset forms. The early-onsetform is also called the infantile form, and the late-onset form can be further divided into juvenile oradult forms. The course of the disease is similar, but the age at which symptoms appear varies, as does
Page 153 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

the rate at which symptoms progress. The age at which symptoms begin is usually similar amongfamily members.
Infantile Form
This is the most common form of MLD, accounting for 50-60% of all cases. Symptoms appearbetween the first and second years of life. Initially, affected children lose any language abilities theyhave developed and have trouble walking. Gradually their muscles waste away and become rigid. Theywill lose mental function and often experience seizures and loss of sensation in their limbs. By the finalstages of the disease, children with infantile MLD become blind and deaf and require a feeding tube.They are unresponsive to their surroundings and become paralyzed. Infantile MLD is usually fatal bythe age of 10.
Juvenile Form
In the juvenile form of MLD, symptoms appear after the age of 3 but before adolescence (12 to 14).Between 20-30% of people with MLD have the juvenile form. Initial signs of the disease are oftendifficulties in school and behavioral problems. Clumsiness, slurred speech, incontinence, and strangebehavior often prompt parents to seek a diagnosis. As the disease continues, symptoms are similar toinfantile MLD, but progress more slowly. The disease is usually fatal 10 or 20 years after symptomsappear.
Adult Form
In the adult form of MLD, symptoms appear after puberty and may not appear until a person’s 40s or50s. Roughly 15 to 20% of people with MLD have the adult form. Early signs of the disease ofteninclude personality changes, problems at school or work, numbness in the extremities of one’s limbs,muscle weakness and loss of coordination, psychiatric problems such as delusions or hallucinations, ordrug and alcohol abuse. It can be initially misdiagnosed as schizophrenia, depression, or multiplesclerosis.
Over time, an affected person’s behavior will grow inappropriate and he or she will have troublemaking good decisions. Everyday skills become difficult and movement will grow spastic andawkward. Eventually a person affected by the adult form will lose the ability to carry on aconversation. In the final stages of the disease, symptoms are similar to the infantile form—blindness,deafness, unresponsiveness, and paralysis.
This form of the disease progresses more slowly than the other forms. Those affected may experienceperiods of stability or periods of particularly rapid decline. People with the adult form of MLD maylive 20 or 30 years after their initial diagnosis.
Page 154 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is metachromatic leukodystrophy?Worldwide, 1 in every 40,000 to 160,000 people have metachromatic leukodystrophy. In certainpopulations, the prevalence can be much higher. These populations include Habbanite Jews in Israel (1in 75), Israeli Arabs (1 in 8,000), Christian Israeli Arabs (1 in 10,000), and those in western portions ofthe Navajo Nation (1 in 2,500).
How is metachromatic leukodystrophy Treated?There is no cure for MLD. Bone marrow transplantation may be an option for some people with MLD.It has shown the most promise in people who are not yet showing symptoms of MLD. At best it slows,but does not stop, the progression of the disease. This is a controversial treatment because of itssubstantial health risks.
Other treatments aim to manage symptoms of the disease as they arise. Seizures and muscle tightnessmay be treated with medication. Physical therapy may help preserve movement as long as possible.Also helpful may be walking aids, wheelchairs, and feeding tubes.
What is the Prognosis for Someone With metachromaticleukodystrophy?All people with metachromatic leukodystrophy will experience mental and motor deterioration,eventually reaching a state of paralysis and unresponsiveness.
Most children with the infantile form die by the age of 10. Those with the juvenile form typicallydevelop symptoms between the ages of 3 and 14 and can live 10 to 20 years after the onset ofsymptoms. The adult form of the disease is more variable, but affected adults may not developsymptoms until their 40s or 50s and can live 20 to 30 years after symptoms begin. Death mostcommonly occurs from pneumonia or other infections.
Page 155 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Mucolipidosis IVGene: MCOLN1
Variant Accuracy Precision511_6944del 1.000 1.000IVS3-2A>G 1.000 1.000
Detection Rate Population<10% African American
96.30% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is mucolipidosis IV?Mucolipidosis IV is a rare inherited condition that affects the development of the nerves. In about 15%of cases, it also causes existing nerves to degenerate. Most infants with the condition are unable to situp, crawl, or control their hand motions. They also chew and swallow very slowly, because themuscles of their mouth and face move slowly or not at all.
Children with the condition never learn to walk independently, although a few have learned to use awalker. When they are able to speak, they tend to do so either very slowly or very quickly and slurwords, mumble, or whisper. In addition, they rarely learn more than a few words, although somechildren with mucolipidosis IV have learned to communicate with a few dozen basic signs. In general,people with the disease only reach a developmental age of 12 to 15 months.
Mucolipidosis IV leads to poor vision caused by cloudy corneas (the clear front part of the eye) anddegeneration of the retina (the part inside the eye which translates light into images). People with thedisease are also prone to dry, irritated eyes, crossed eyes, and pupils that respond slowly to changes inlight levels. Although infants with the condition may be born with nearly normal vision, their visionalmost always starts to deteriorate by the age of 5. Virtually everyone with the condition is severelyvisually impaired by the early teens.
Page 156 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

About 5% of people with the condition have an atypical variation with less severe movement andvision problems.
How Common is mucolipidosis IV?Fewer than 100 cases of mucolipidosis IV have been reported in medical literature. More than 80% ofthose affected are of Ashkenazi Jewish background, and the disease is rare outside this population.Roughly 1 in 100 Ashkenazi Jews is a carrier.
How is mucolipidosis IV Treated?Treatment for mucolipidosis IV focuses on ensuring comfort and improving function. Physical therapy,foot and ankle orthotics, walkers, and wheelchairs can help maximize mobility. Speech therapy mayimprove the ability to communicate. Younger children frequently develop eye irritation, whichlubricating eye drops, gels, or ointments can soothe.
What is the Prognosis for Someone With mucolipidosisIV?Mucolipidosis IV typically shortens one's lifespan, but people with the disease commonly reachadulthood and some are known to be alive in their mid-40s.
Page 157 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Muscle-Eye-Brain DiseaseGene: POMGNT1
Variant Accuracy PrecisionIVS17+1G>A 1.000 1.000
Detection Rate Population27.70% African American27.70% Ashkenazi Jewish27.70% Eastern Asia27.70% Finland27.70% French Canadian or Cajun27.70% Hispanic27.70% Middle East27.70% Native American27.70% Northwestern Europe27.70% Oceania27.70% South Asia27.70% Southeast Asia27.70% Southern Europe
What is muscle-eye-brain disease?Muscle-eye-brain disease (MEB) is an inherited condition causing a number of symptoms includingmuscle weakness, vision abnormalities, brain structure abnormalities, and severe mental disability.
MEB causes congenital muscular dystrophy, a form of muscle weakness that is present from birth ordevelops shortly after birth. It causes an infant to feel floppy in all of his or her muscles, includingthose of the face. He or she may also exhibit involuntary muscle jerks or twitches.
Eye problems associated with MEB include severe near-sightedness and glaucoma, among others.
Another hallmark of MEB is a brain abnormality known as cobblestone lissencephaly (or type IIlissencephaly). The brain develops a bumpy “cobblestone” appearance and lacks the normal foldingstructure. Other structural changes in the brain are also present. Children with MEB may have abuildup of fluid around the brain that can create a dangerous amount of pressure.
The severity of symptoms can vary among people with MEB.
How Common is muscle-eye-brain disease?MEB is very rare, although its exact prevalence is unknown.
Page 158 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is muscle-eye-brain disease Treated?There is no successful treatment or cure for MEB. Medical specialists can help treat specificsymptoms, such as using medication to control seizures, physical and occupational therapy to aid inmovement, and special eye glasses to help make the most of the child's vision.
What is the Prognosis for Someone With muscle-eye-brain disease?The prognosis for a person with MEB varies depending on the severity of the symptoms, but isgenerally poor. Studies have shown people with MEB typically die between the ages of 6 and 16.
Page 159 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

MYH-Associated PolyposisGene: MUTYH
Variant Accuracy PrecisionY165C 1.000 1.000
Detection Rate Population23.10% African American23.10% Ashkenazi Jewish23.10% Eastern Asia23.10% Finland23.10% French Canadian or Cajun23.10% Hispanic23.10% Middle East23.10% Native American23.10% Northwestern Europe23.10% Oceania23.10% South Asia23.10% Southeast Asia23.10% Southern Europe
What is MYH-associated polyposis?MYH-associated polyposis (MAP) is an inherited condition that causes abnormal, mushroom-likegrowths in the large intestine. People with MAP face a significantly increased risk of developingcolorectal cancer relatively early—often between their 20s and 50s.
People with MAP typically develop between 3 and 100 polyps in their large intestine, and occasionallyin their rectum or small intestine. These polyps are benign (not cancerous), but can become cancerousover time. Polyps often begin to develop in the late teens or early 20s. Rarely does the disease createsymptoms in children.
Unfortunately the exact cancer risks associated with MAP are not yet well understood. There are noavailable statistics indicating how many people with MAP develop colon cancer, nor the average age atwhich that happens. There are ongoing studies to determine whether being a carrier for MAP increasesone's chances of developing colorectal cancer, however findings to date are conflicting.
How Common is MYH-associated polyposis?Studies have estimated that 1 in 100 Caucasians are carriers of MAP. Because the genetic basis for thedisease has only recently come to light, statistics about other populations are unknown. The percentageof colon cancer caused by MAP is also unknown.
Page 160 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is MYH-associated polyposis Treated?People with MAP should undergo regular colonoscopies to screen for cancer. Some expertsrecommend a colonoscopy every 2 to 3 years beginning at the age of 18 to 20. Other tests are alsorecommended to check the small intestine periodically.
What is the Prognosis for Someone With MYH-associatedpolyposis?Because the genetic basis of the disease is newly discovered and not well understood, there is no singleprognosis for a person with MAP. It is not known how many will develop colon cancer. It should benoted that if caught early, colon cancer is treatable.
Page 161 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Niemann-Pick Disease Type AGene: SMPD1
Variant Accuracy PrecisionfsP330 1.000 1.000L302P 1.000 1.000R496L 1.000 1.000
Detection Rate Population<10% African American
95.00% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Niemann-Pick disease type A?Niemann-Pick disease type A is an inherited disease in which the body cannot properly metabolizecertain fats due to a deficient enzyme called acid sphingomyelinase. This leads to an excess ofcholesterol and fat in the body. The disease causes mental disability, loss of motor skills, andenlargement of the liver and spleen, among other symptoms. The disease is often fatal by the age of 2or 3.
Symptoms of Niemann-Pick disease type A usually begin within the first few months of life. By theage of six months, infants with the disease have difficulty feeding, display an enlarged abdomen, andwill begin to lose the motor skills they have developed. Seizures and spastic movement are common.Most will not learn to sit independently, crawl, or walk. They have poor muscle tone and developcherry-red spots in their eyes. Many have a yellow tinge to the skin and whites of the eye (jaundice).
Intellectual and motor skills will progressively and rapidly decline. These children may showvomiting, irritability, lung infections, and difficulty sleeping.
Page 162 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Niemann-Pick disease type A?Niemann-Pick disease type A is a rare disease, occurring most frequently in Ashkenazi Jews, amongwhom 1 in 100 is a carrier. The disease is not limited to Ashkenazi Jews, however, and has occurred inpeople of all ethnicities. Type A is the most common form of Niemann-Pick disease, accounting for85% of cases.
How is Niemann-Pick disease type A Treated?Unfortunately there are no effective treatments for Niemann-Pick disease type A. Medicalprofessionals can attempt to treat the symptoms through physical therapy, monitoring of nutrition, andmedication to help sleep disorders. Such treatment cannot stop the decline caused by the disease,however.
What is the Prognosis for Someone With Niemann-Pickdisease type A?The prognosis for a person with Niemann-Pick disease type A is poor. It is a severe disease which istypically fatal by the age of 2 or 3.
Page 163 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Niemann-Pick Disease Type CGene: NPC1
Variant Accuracy PrecisionI1061T 1.000 1.000
Detection Rate Population17.80% African American
<10% Ashkenazi Jewish17.80% Eastern Asia
<10% Finland<10% French Canadian or Cajun
17.80% Hispanic17.80% Middle East17.80% Native American
<10% Northwestern Europe17.80% Oceania17.80% South Asia17.80% Southeast Asia
<10% Southern Europe
What is Niemann-Pick disease type C?Niemann-Pick disease type C is an inherited condition in which the body cannot properly metabolizecholesterol and fats, resulting in an excess of these substances in the body. Cholesterol buildup in theliver causes severe liver disease, and fat accumulation in the brain leads to learning disabilities andprogressive neurological symptoms.
The first symptom of the disease, which can appear at any age from infants to adults, is an enlargedliver, enlarged spleen, or jaundice. In some cases, it is possible to detect the disease in an unborn childvia ultrasound, but the disease is most commonly diagnosed in school-aged children. Symptoms mayinclude sudden muscle problems such as seizures, clumsiness, tremors, problems walking, suddenfalls, slurred speech, and trouble moving the eyes up and down. As the condition progresses, thesechildren develop learning disabilities, psychological problems, or even dementia, and often lose theability to speak. Eventually, people with Niemann-Pick disease type C lose the ability to move theirfacial muscles or swallow, making feeding through a stomach tube necessary.
For those diagnosed during childhood, the disease is usually fatal in the late teens or twenties due topneumonia. People diagnosed in adulthood generally survive 10 to 20 years after diagnosis.
At the cellular level, Niemann-Pick disease type C can be caused by two different genetic mutations.Type C1 is caused by a mutation in the NPC1 gene, and type C2 is caused by a mutation in the NPC2gene. Although the genetic mutations are different, the resulting symptoms are the same because NPC1
Page 164 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

and NPC2 must work together to remove cholesterol and lipids from body cells. Of the known cases ofNiemann-Pick disease type C, 95% have been type C1 while 5% have been C2.
How Common is Niemann-Pick disease type C?Niemann-Pick disease type C is estimated to affect 1 in 150,000 people. It is more common amongFrench Acadians in Nova Scotia, people of Hispanic descent in specific parts of Colorado and NewMexico, and a small Bedouin group in Israel.
How is Niemann-Pick disease type C Treated?At this time, there is no cure for Niemann-Pick disease type C. Treatment focuses on managingsymptoms with medication for seizures, sedatives for sleep disturbances, physical therapy to maintainmobility, and speech therapy to preserve communication as long as possible. Chest physiotherapy andantibiotics may help to prevent regular lung infections. People with the condition need a gastronomytube for feeding when they can no longer swallow well enough to avoid choking or malnutrition.
What is the Prognosis for Someone With Niemann-Pickdisease type C?Niemann-Pick disease type C is usually fatal 10 to 20 years after diagnosis. In children who showsymptoms at an early age, disease progression is usually faster compared to people whose symptomsappear later in life. Over half of people with this disease will be diagnosed by the age of 10.
Page 165 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Nijmegen Breakage SyndromeGene: NBN
Variant Accuracy Precision657del5 1.000 1.000
Detection Rate Population<10% African American
78.00% Ashkenazi Jewish<10% Eastern Asia
78.00% Finland78.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
78.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
78.00% Southern Europe
What is Nijmegen breakage syndrome?Nijmegen breakage syndrome (NBS) is an inherited disease in which the body’s DNA is prone tobreakages. People with NBS often develop cancer at an early age and experience frequent lung andsinus infections. They also show intellectual decline, eventually leading to mild-to-moderate mentaldisability. People with NBS can live into adulthood, but typically not beyond their 30s or 40s.
Infants with NBS are often born with large heads. Their physical growth is often slow, leaving themsmaller than average for their age. They have characteristic features, including a sloping forehead,small chin, big ears, and prominent nose, which become more apparent later in childhood.
In one study, 35% of the 70 people studied who had NBS developed cancer—most commonly a typeknown as B-cell lymphoma—between the ages of 1 and 34. People with NBS cannot tolerate the highdoses of ionizing radiation often used to treat cancer, and must find alternate treatment methods.
Immunodeficiency—the reduced ability of the body to fight off infection—is another symptomfrequently associated with NBS. As a result, the disease causes frequent infections in the lungs, ears,sinuses, and urinary tract. Recurrent bronchitis can be life-threatening.
Intellect appears to develop normally or near-normally in early childhood, but typically declines untilthe person reaches mild-to-moderate levels of mental disability around the age of 10.
Page 166 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Nijmegen breakage syndrome?Scientists estimate that 1 in 100,000 births is affected by NBS. The disease is most common in peopleof Eastern European or Slavic background, specifically those from Poland, the Czech Republic, andthe Ukraine. There the carrier frequency may reach as high as 1 in 155. More than 40 cases of NBShave been diagnosed in North America.
How is Nijmegen breakage syndrome Treated?There is no treatment to address the underlying cause of NBS, but certain symptoms can be treated.
Vitamin E and folic acid supplements may be helpful. In some people, intravenous infusions withimmunoglobulin may help reduce infections. Preventative antibiotics are another option for treatinginfection. Special education and speech therapy can be helpful as well.
Large doses of radiation must be avoided in people with NBS, even before birth. Cancer treatmentstherefore must be adapted.
What is the Prognosis for Someone With Nijmegenbreakage syndrome?Some people with NBS do live into adulthood, though typically the lifespan does not extend beyondone's 30s or 40s. The longest known lifespan of a person with NBS is 53. Cancer is the most commoncause of death among people with NBS, followed by lung infections leading to respiratory failure.
Page 167 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Northern EpilepsyGene: CLN8
Variant Accuracy PrecisionR24G 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia>99% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Northern epilepsy?Northern epilepsy is an inherited disease that causes seizures and leads to severe mental disability inadulthood.
Children with Northern epilepsy appear normal until they begin to develop seizures between the agesof 5 and 10. The seizures increase in frequency until puberty, after which they decline in frequency,but do not disappear. Within 2 to 5 years of the first seizure, the child begins to decline mentally. Thisdecline is progressive and by the age of 30, the person is mentally disabled (an IQ less than 70),regardless of whether the seizures were controlled by medication. After age 30, people with the diseasemay also become clumsy in their movements and have problems with balance.
One third of people with the disease will also develop mild loss of visual acuity in adulthood.
Northern epilepsy belongs to a heterogenous group of diseases known as neuronal ceroidlipofuscinoses. Northern epilepsy is caused by a mutation in the gene CLN8. This gene carries theinstructions for a particular protein, but the function of this protein is unknown.
Page 168 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Northern epilepsy?In Finland, 1 in 135 people are carriers of the disease. All cases to date have been among Finnishpeople, particularly in the northern part of the country. Fewer than 50 cases have been described in themedical literature.
How is Northern epilepsy Treated?There is no effective treatment for Northern epilepsy. Seizures can be controlled with medication,however this will not slow the progression of the disease towards mental disability.
What is the Prognosis for Someone With Northernepilepsy?People with Northern epilepsy often live to the age of 50 or 60, but have significant mental andphysical impairments for much of their lives.
Page 169 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Pendred SyndromeGene: SLC26A4
Variant Accuracy PrecisionL236P 1.000 1.000E384G 1.000 1.000T416P 1.000 1.000
Detection Rate Population14.60% African American44.00% Ashkenazi Jewish14.60% Eastern Asia44.00% Finland44.00% French Canadian or Cajun14.60% Hispanic14.60% Middle East14.60% Native American44.00% Northwestern Europe14.60% Oceania14.60% South Asia14.60% Southeast Asia44.00% Southern Europe
What is Pendred syndrome?Pendred syndrome is an inherited condition that affects the body's ability to make a protein calledpendrin, which is important for normal functions of the inner ear and thyroid.
People with the condition are usually born severely to profoundly deaf, although some lose theirhearing rapidly in infancy or early childhood and others have only moderate hearing loss that does notworsen over time. The inner ear malformations that are typical of Pendred syndrome may also causebalance problems.
Affected individuals may develop a goiter, a large swelling at the base of the neck caused by thyroidenlargement. This symptom usually appears several years after hearing loss is diagnosed. It can happenat any time during late childhood, adolescence, or adulthood. Pendred syndrome does not usuallyaffect thyroid function, however goiters can put pressure on the esophagus and windpipe, interferingwith swallowing and breathing.
How Common is Pendred syndrome?The frequency of Pendred syndrome is unknown, but some researchers believe it is responsible for 1 in10 infants who are born deaf.
Page 170 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is Pendred syndrome Treated?Treatment for Pendred syndrome focuses on addressing hearing loss. Children with the conditionshould be fitted for hearing aids early in life. Cochlear implants show some promise for restoring somehearing to people who are severely to profoundly deaf. Children should receive special educationalprograms for the hearing-impaired.
For those who develop goiters large enough to cause breathing or swallowing difficulties, treatmentmay include radioactive iodine to shrink the swelling or surgery to remove all or part of the thyroid.
What is the Prognosis for Someone With Pendredsyndrome?Pendred syndrome causes moderate to profound hearing loss, but does not affect lifespan.
Page 171 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Phenylalanine Hydroxylase DeficiencyGene: PAH
Variant Accuracy PrecisionIVS12+1G>A 1.000 1.000L48S 1.000 1.000R158Q 1.000 1.000R252W 1.000 1.000G272X 1.000 1.000R408Q 1.000 1.000Y414C 1.000 1.000IVS-10int-546 1.000 1.000I65T 1.000 1.000R261Q 1.000 1.000R408W 1.000 1.000
Detection Rate Population34.10% African American34.10% Ashkenazi Jewish34.10% Eastern Asia34.10% Finland34.10% French Canadian or Cajun34.10% Hispanic34.10% Middle East34.10% Native American34.10% Northwestern Europe34.10% Oceania34.10% South Asia34.10% Southeast Asia34.10% Southern Europe
What is phenylalanine hydroxylase deficiency?Phenylalanine hydroxylase deficiency is a treatable inherited disease in which the body cannotproperly process the amino acid phenylalanine due to a deficient enzyme called phenylalaninehydroxylase. If severe forms of the disease go untreated, the buildup of phenylalanine can be toxic tothe brain, causing impaired development and leading to severe and irreversible mental disability. Iftreated early and consistently however, people with phenylalanine hydroxylase deficiency can leadcompletely normal lives.
The disease can be divided into several categories based on the amount of enzyme deficiency: Classicphenylketonuria (PKU), variant PKU, and non-PKU hyperphenylalaninemia (non-PKU HPA). Sincethe mid-1960s, it has been standard for hospitals in North America to screen newborns for
Page 172 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

phenylalanine hydroxylase deficiency using a drop of blood obtained from a heel prick. This is now aroutine practice in most developed countries.
It can be difficult to predict how severely affected a child will be based on the particular geneticmutations they carry. Children with any form phenylalanine hydroxylase deficiency should beevaluated by a specialist immediately after birth.
Classic PKU
Classic PKU is the most common and severe form, resulting from an absence or near absence of thephenylalanine hydroxylase enzyme.
If PKU is not promptly diagnosed and treated with a special diet, mental disability will occur, alongwith a number of other symptoms including a small head, seizures, behavior problems, a "mousy" or"musty" odor, abnormal gait, low bone density, and eczema (a skin condition). These are all avoidableif the proper diet is instituted shortly after birth.
Variant PKU
Variant PKU is an intermediate form of the disease, less severe than classic PKU but more severe thannon-PKU HPA. A child with variant PKU is at risk for developing the symptoms associated withclassic PKU. Though the symptoms may be milder, there is still a risk for impaired mentaldevelopment if the child's intake of phenylalanine is not monitored.
Non-PKU Hyperphenylalaninemia
Non-PKU HPA is the mildest form of phenylalanine hydroxylase deficiency. People with non-PKUHPA have a higher level of phenylalanine hydroxylase than do people with classic or variant PKU andare consequently at lower risk for developing brain damage. Some people with non-PKU HPA are ableto tolerate a normal diet and do not require treatment. This will vary from person to person and must bedetermined by a medical professional based on the levels of phenylalanine in the person’s blood.
How Common is phenylalanine hydroxylase deficiency?The frequency of carriers and affected individuals in select populations is listed below.
Ethnic Group Carrier Rate Affected Rate
Turkish 1 in 26 1 in 2,600
Irish 1 in 33 1 in 4,500
Caucasian American 1 in 50 1 in 10,000
Page 173 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Ethnic Group Carrier Rate Affected Rate
East Asian 1 in 51 1 in 10,000
Finnish 1 in 200 1 in 160,000
Japanese 1 in 200 1 in 160,000
Ashkenazi Jewish 1 in 225 1 in 200,000
How is phenylalanine hydroxylase deficiency Treated?The degree of enzyme deficiency varies among people with phenylalanine hydroxylase deficiency, andtherefore the treatment must also be individualized based on the levels of phenylalanine in the blood.An infant with any form of phenylalanine hydroxylase deficiency should be evaluated immediatelyafter birth to determine whether or not he or she requires treatment. A blood test can reveal the amountof functioning phenylalanine hydroxylase in the body and this will indicate the amount ofphenylalanine the person can safely consume.
While people with classic PKU must adhere to a strict low-phenylalanine diet, others with milder formof the disease are able to safely consume small amounts of the amino acid. For people with non-PKUHPA, treatment may not even be necessary.
Generally speaking, a diet low in protein and free from phenylalanine is important in preserving mentalfunction in a person with classic PKU. Phenylalanine-free formulas are available for infants.Maintaining appropriate levels of phenylalanine in the brain can be achieved through blood testing anddiet adjustment. This must be closely supervised by medical professionals. In most cases, this specialdiet must be maintained for life.
People with any form of phenylalanine hydroxylase deficiency should be conscious to avoidconsuming aspartame, an artificial sweetener that contains phenylalanine.
Women with phenylalanine hydroxylase deficiency who become pregnant must be particularly carefulto maintain safe levels of phenylalanine in their own bodies in order to avoid birth defects in theirchildren. Ideally this begins prior to conception.
In late 2007, the medication sapropterin dihydrochloride (brand name: Kuvan) was approved by theFDA for use in people with phenylalanine hydroxylase deficiency. In some, it can enhance the activityof the deficient enzyme and lower levels of phenylalanine in the body, allowing for a relaxation of thedietary restrictions. Some people with the disease do not respond to the drug, however. The peoplewho do respond to this treatment usually have milder forms of the disease.
Page 174 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With phenylalaninehydroxylase deficiency?If a person with classic or variant PKU is treated early and consistently for the disease, the prognosiscan be excellent. Many people with PKU have gone on to lead normal lives with normal intelligenceand lifespan.
If treatment is not begun early or adequately maintained, a person with a more severe form of PKU isat risk for severe and irreversible brain damage.
The prognosis is good for a person with non-PKU HPA. He or she may lead a normal life withouttreatment.
Page 175 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Polyglandular Autoimmune SyndromeType 1Gene: AIRE
Variant Accuracy PrecisionY85C 1.000 1.000R257X 1.000 1.000
Detection Rate Population<10% African American
23.63% Ashkenazi Jewish<10% Eastern Asia
89.00% Finland23.63% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
23.63% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
23.63% Southern Europe
What is polyglandular autoimmune syndrome type 1?Polyglandular Autoimmune Syndrome Type 1 (PAS-1) is an inherited disease in which the body’simmune system mistakenly attacks healthy cells, notably the glands that produce the body’s hormones.People with PAS-1 have at least two of the disease’s main symptoms: fungal infections of the skin andmucous membranes, decreased function in the parathyroid glands, and decreased function in theadrenal glands (Addison disease). Many people with the disease have all three main symptoms. Thereare also numerous and diverse other symptoms which can occur.
In the majority of people with PAS-1, the first symptom to appear is recurrent and persistent fungalinfections of the skin and mucous membranes, such as the in the moist lining of the nose and mouth.These infections typically begin between the ages of 3 and 5, although they can occur any time beforeone’s 30s.
Frequently the second symptom of the disease to appear is an underactive parathyroid gland(hypoparathyroidism). This typically occurs before the age of 10. The parathyroid glands normallysecrete a hormone used to regulate the amount of calcium and phosphorous in the bone and blood. Anunderactive parathyroid gland can cause numerous symptoms including tingling in the lips, fingers,
Page 176 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

and toes; muscle cramps; pain in the abdomen, face, legs, and feet; weakness or fatigue; and dry hairand skin.
Often the third symptom to appear is underactive adrenal glands, a condition known as Addisondisease. This disease typically appears before the age of 15. Addison disease can cause fatigue, muscleweakness, weight loss, low blood pressure, and changes in skin coloration.
There are numerous other symptoms which can also occur in people with PAS-1. These includechronic liver disease, extreme fatigue due to a problem with red blood cells, skin disease, total bodyhair loss, an underactive pituitary gland, abnormalities in the ovaries and testes, diarrhea, difficultyabsorbing nutrients from food, and eye problems, among others.
The most common pattern with PAS-1 is that the three main symptoms of the disease—fungalinfections of the skin, underactive parathyroid gland, and Addison disease—develop in that orderbefore the age of 20. The other symptoms associated with the disease may then begin sporadically overtime until one’s 50s, when the symptoms typically stabilize. This does not hold true for all people withPAS-1, however. Generally speaking, the earlier in life that the main symptoms appear, the more likelyit is that additional symptoms will develop.
How Common is polyglandular autoimmune syndrometype 1?PAS-1 is a rare condition in the United States, but it is more common among certain ethnic groups.The number of people affected include:
• Iranian Jews: 1 in 6,500 to 9,000• Sardinians: 1 in 14,000• Finns: 1 in 25,000• Slovenians: 1 in 43,000• Norwegians: 1 in 80,000 to 90,000• Poles: 1 in 129,000
How is polyglandular autoimmune syndrome type 1Treated?There is no cure for PAS-1. Each symptom must be treated as it arises and lifelong regular checkupsare necessary to look for any new symptoms. It is important to discover and treat new symptoms assoon as possible to prevent permanent damage to the body.
Physicians often prescribe hormone replacement therapy or intravenous steroids for people withPAS-1. Calcium and vitamin D are often helpful to treat an underactive parathyroid gland. Fungalinfections can be treated with medication.
Page 177 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Other symptoms are treated as they arise. For example, people with diabetes can take insulin andmonitor their diet.
What is the Prognosis for Someone With polyglandularautoimmune syndrome type 1?The prognosis for a person with PAS-1 varies depending on the number and severity of his or hersymptoms. Early detection of the disease and its component symptoms is important for preventing life-threatening scenarios. With careful monitoring, it is possible to have a normal or near-normal lifespan.Women with PAS-1 can give birth, and men with PAS-1 can father healthy children.
Page 178 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Pompe DiseaseGene: GAA
Variant Accuracy PrecisionD645E 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Pompe disease?Pompe disease, also known as glycogen storage disease type II, is an inherited disorder whose primarysymptom is progressive weakness in the muscles used for mobility and breathing. In infants withPompe disease, the heart muscles are often severely affected as well. These symptoms are caused by amutation in an enzyme called alpha-glucosidase (also called maltase) that breaks down glycogen, astored form of sugar used for energy. As a result, glycogen builds up in the body, notably in themuscles, and damages individual cells.
There are two main types of Pompe disease: infantile-onset and late-onset forms. The severity ofsymptoms, age at which symptoms begin, and rates of disease progression are related to the degree ofalpha-glucosidase deficiency.
Infantile-onset Form
The infantile form is the most common and most severe type of Pompe disease. Babies with thedisease may appear normal at birth, but begin to show symptoms in the first few months of life. Theydevelop general muscle weakness and poor muscle tone, which causes their bodies to seem limp asthey are unable to move, hold up their heads, or feed. They fail to gain weight and grow at theexpected rate. Breathing problems can be compounded by lung infections. These infants have enlargedhearts and livers, and many also have enlarged tongues. The disease progresses rapidly and most
Page 179 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

infants with Pompe disease will die within the first year of life, often from heart or lung failure. Inpeople with the infantile form of the disease, alpha-glucosidase is either entirely missing or inactive.
Late-onset Form
The late onset form of Pompe disease is due to a partial deficiency in alpha-glucosidase. Symptomscan begin at any time, from childhood to adulthood. In this form of the disease, muscle weaknesseventually leads to breathing problems and death from lung failure. The heart may be involved, but itwill not be enlarged. These people will lose mobility and eventually require a wheelchair or becomebedridden. Machines may become necessary in order to breathe.
This form of the disease progresses more slowly, and life expectancy is better than in the infantile-onset form. People who develop symptoms of Pompe disease in late childhood often die in their 20sand 30s. Those who develop symptoms later may experience a slower progression, but unfortunatelytheir lifespan will also be curtailed.
How Common is Pompe disease?Pompe disease affects roughly 1 in 100,000 people. The infantile-onset form is the most common typeof Pompe disease.
How is Pompe disease Treated?In 2006, the FDA approved an enzyme replacement therapy called Myozyme for people with Pompedisease. Myozyme has been shown to decrease heart size, maintain normal heart function, and improvemuscle tone and strength in people with the infantile-onset form of the disease. It is too soon to gaugehow it will affect people with the disease long-term.
Adults and children with Pompe disease are often prescribed a protein-rich diet and a daily exerciseregimen to help muscle tone and strength. They must also carefully monitor and treat lung infections.
What is the Prognosis for Someone With Pompe disease?Babies born with the infantile-onset form of Pompe disease typically die within the first year of life,though enzyme replacement therapy can now prolong that lifespan. For people with the late-onsetforms of the disease, lifespan will depend upon the age at which symptoms begin and the degree ofalpha-glucosidase impairment. In general, the later in life symptoms develop, the slower they willprogress. Unfortunately, this disease will greatly curtail the lifespan of those affected. Most peoplewith Pompe disease will die from lung failure.
Page 180 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

PPT1-Related Neuronal CeroidLipofuscinosisGene: PPT1
Variant Accuracy PrecisionL10X 1.000 1.000T75P 1.000 1.000R122W 1.000 1.000R151X 1.000 1.000
Detection Rate Population57.55% African American57.55% Ashkenazi Jewish57.55% Eastern Asia98.00% Finland57.55% French Canadian or Cajun57.55% Hispanic57.55% Middle East57.55% Native American57.55% Northwestern Europe57.55% Oceania57.55% South Asia57.55% Southeast Asia57.55% Southern Europe
What is PPT1-related neuronal ceroid lipofuscinosis?PPT1-related neuronal ceroid-lipofuscinosis (NCL) is an inherited disease that causes degeneration ofthe brain leading to a progressive loss of mental and motor skills. It can also cause blindness, andtypically leads to an early death. In the final stages of the disease, affected individuals will bemotionless and in a vegetative state.
There are several forms of NCL, largely differentiated by the gene responsible and the age at whichsymptoms begin. Mutations in the PPT1 gene typically result in the infantile or juvenile form of NCL.
Infantile Form
The infantile form of NCL (INCL) usually begins to cause noticeable symptoms between the ages of 6months and 24 months. Initially, infants will show developmental delays and experience seizures orjerking movements. Often these infants will have small heads. Blindness and seizures will be presentby 24 months, after which mental functions will deteriorate. The child’s movement will become spasticand uncontrolled and he or she will develop dementia.
Page 181 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Juvenile Form
The symptoms of juvenile NCL (JNCL), also called Batten disease, often begin between the ages of 4and 10. These children rapidly lose their vision, which is often the first noticeable symptom. Theytypically become completely blind within two years. People with JNCL often develop periodic seizuresbetween the ages of 5 and 18.
Between the ages of 8 and 14, mental functions typically decline. Children may have difficulty withspeech and show behavioral problems. Some people with JNCL also develop psychiatric problemsincluding disturbed thoughts, attention problems, and aggression. These problems can eventuallyprogress to dementia.
People with JNCL also show a decline in motor function and may have difficulty controlling their ownmovement.
How Common is PPT1-related neuronal ceroidlipofuscinosis?Approximately 1 in 25,000 people globally are affected by some form of NCL. These diseases aremost common in Scandinavian countries, but occur elsewhere as well. In the United States, anestimated 25,000 families are affected by some form of NCL.
INCL is most common in Finland, where 1 in 20,000 births is affected by the disease and 1 in 70people is a carrier. About half the world’s cases of INCL are in Finland.
In Iceland, 7 in 100,000 births are affected by JNCL. Other countries experience fewer cases of JNCL.One study showed 0.7 cases per 100,000 births in Germany.
Exactly how many cases of NCL are caused by mutations in the PPT1 gene is unknown.
How is PPT1-related neuronal ceroid lipofuscinosisTreated?There is no treatment for the underlying cause of NCL. Treatments can only address the symptoms asthey arise. Various medications can be useful for treating seizures, poor muscle tone, sleep disorders,mood disorders, excessive drooling, and digestion. In some people, a feeding tube is also helpful.
Page 182 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With PPT1-relatedneuronal ceroid lipofuscinosis?The prognosis for a person with NCL depends upon the type of the disease he or she has. People withINCL or JNCL will become blind and will deteriorate mentally. They will eventually enter a vegetativestate and become totally dependent on others to care for them.
Among those with INCL, death usually occurs in childhood.
Among those with JNCL, death usually occurs between one's late teens to 30s.
Page 183 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Primary Hyperoxaluria Type 1Gene: AGXT
Variant Accuracy PrecisionF152I 1.000 1.000G170R 1.000 1.000I244T 1.000 1.000
Detection Rate Population33.99% African American33.99% Ashkenazi Jewish33.99% Eastern Asia33.99% Finland33.99% French Canadian or Cajun33.99% Hispanic33.99% Middle East33.99% Native American33.99% Northwestern Europe33.99% Oceania33.99% South Asia33.99% Southeast Asia33.99% Southern Europe
What is primary hyperoxaluria type 1?Primary hyperoxaluria type 1 (PH1) is an inherited disease in which the lack of a particular liverenzyme causes the body to accumulate excess amounts of a substance called oxalate. This oxalateleads to a buildup of insoluble calcium salts in the kidneys and other organs. If untreated, it results inlife-threatening kidney failure.
People with PH1 are prone to recurrent kidney stones that can lead to kidney failure. Among peoplewith PH1, 50% experience kidney failure by the age of 15, 80% by the age of 30.
In addition to the kidneys, PH1 also leaves insoluble calcium deposits in other body tissues. This canlead to bone pain; vision loss; tingling, numbness, or pain in the extremities; enlargement of the liverand spleen; and problems with the electrical system of the heart (heart block).
Symptoms typically begin between the ages of 1 and 25, with roughly 80% showing signs of thedisease in late childhood or early adolescence. Another 10% of people with PH1 show symptoms inearly infancy (before the age of 6 months) while the remaining 10% do not show symptoms until their40s or 50s.
Page 184 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is primary hyperoxaluria type 1?PH1 is quite rare, with estimates placing its frequency worldwide between 1 in 100,000 and 1 in1,000,000. In Europe, it affects an estimated 1 in 120,000 people. It is thought to be more common inTunisia, Iran, and Israeli Arab and Druze populations.
How is primary hyperoxaluria type 1 Treated?About half of people with PH1 respond to high doses of vitamin B6, which can reduce the amount ofoxalate in the body. In some of these people, the oxalate level can be normalized, while in others it ismerely reduced to a healthier level.
People with the condition should drink plenty of water. A physician may prescribe medication or othervitamins to help lower oxalate levels and inhibit the formation of kidney stones.
One important option in treating PH1 is organ transplantation of the liver and kidneys. Because adeficient liver enzyme leads to kidney failure, early liver transplantation may avoid the need to alsotransplant new kidneys. Kidney replacement alone is not a sufficient treatment as the liver coulddestroy the new kidneys as well.
People with PH1 should avoid extremely large doses of vitamin C as well as foods high in oxalate,including chocolate, rhubarb, and starfruit.
What is the Prognosis for Someone With primaryhyperoxaluria type 1?The prognosis for a person with PH1 is variable and depends on how early the disease is detected andtreated. Among people with PH1, 50% experience kidney failure by the age of 15, 80% by the age of30. In more severe cases, children will develop kidney failure between the ages of 3 and 6 and willrequire organ transplants in order to survive. Early detection and treatment with vitamin B6 can helpavoid kidney failure in some cases.
Following organ transplant, some people have lived normal or near-normal lifespans.
Women with PH1 have successfully given birth to healthy babies following a liver/kidney transplant.
Page 185 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Primary Hyperoxaluria Type 2Gene: GRHPR
Variant Accuracy Precision103delG 1.000 1.000
Detection Rate Population<10% African American
37.00% Ashkenazi Jewish<10% Eastern Asia
37.00% Finland37.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
37.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
37.00% Southern Europe
What is primary hyperoxaluria type 2?Primary hyperoxaluria type 2 (PH2) is an inherited disease in which the lack of a particular liverenzyme causes the body to accumulate excess amounts of a substance called oxalate. This oxalateleads to a buildup of insoluble calcium salts in the kidneys and other organs. If untreated, it can resultin life-threatening kidney failure.
People with PH2 are prone to recurrent kidney stones that can lead to kidney failure. The disease hassimilar symptoms to primary hyperoxaluria type 1 (PH1), but PH2 tends to be a less aggressive form ofthe disease, even when symptoms start early in life. PH1 and PH2 are caused by different missing liverenzymes.
In addition to the kidneys, PH2 also leaves insoluble calcium deposits in other body tissues, causingproblems with bones, eyes, teeth, nerves, and the heart.
Symptoms typically begin between the ages of 1 and 25, with roughly 80% showing signs of thedisease in late childhood or early adolescence. Another 10% of people with PH2 show symptoms inearly infancy (before the age of 6 months) while the final 10% do not show symptoms until their 40s or50s.
Page 186 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is primary hyperoxaluria type 2?PH2 is very rare, though its exact incidence is unknown. As of 2002, only 37 individuals with thedisease have been described in medical literature.
How is primary hyperoxaluria type 2 Treated?People with the condition should drink plenty of water. A physician may prescribe medications orother vitamins to help lower oxalate levels and inhibit the formation of kidney stones.
While people with PH2 are less likely to develop kidney failure than people with PH1, organtransplantation remains an option if kidney failure does occur. Because a deficient liver enzyme leadsto kidney failure, early liver transplantation may avoid the need to also transplant new kidneys. Kidneyreplacement alone is not a sufficient treatment as the liver could destroy the new kidneys as well.
People with PH2 should avoid extremely large doses of vitamin C as well as foods high in oxalate,including chocolate, rhubarb, and starfruit.
What is the Prognosis for Someone With primaryhyperoxaluria type 2?The prognosis for a person with PH2 is variable and depends on how early the disease is detected andtreated. Some people with the disease will develop kidney failure that may require liver and kidneytransplantation. In other cases, hydration and medication will be sufficient to control the disease.
Generally, people with PH2 have a better long-term outcome than people with PH1 and they requirefewer surgeries.
Following organ transplant, some people have lived normal or near-normal lifespans.
Page 187 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

PycnodysostosisGene: CTSK
Variant Accuracy PrecisionX330W 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is pycnodysostosis?Pycnodysostosis (PYCD), also spelled pyknodysostosis, is an inherited disease that causes the bones tobe abnormally dense while also creating certain bone deformities.
The bones of people with PYCD are brittle and can break easily, even under minimal stress. Fracturesare common in affected people's legs, feets, jaws, and collar bone.
People with the disease are abnormally short—less than five feet in height and often much shorter.They are susceptible to a curved spine (scoliosis), and the collarbone is often deformed as well. Peoplewith PYCD also have significantly shorter finger bones and their nails are small or absent.
People with pycnodysostosis often have characteristic features including a prominent nose, protrudingforehead, and small jaw. Their teeth can be late in coming in, may be missing or irregular, and areprone to cavities. The skull is typically deformed, with the “soft spot” on the top of the head failing toclose.
Many scientists believe that 19th-century French painter Henri de Toulouse-Lautrec had PYCD,though the disease was unknown at the time.
Page 188 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is pycnodysostosis?The frequency of PYCD is unknown, however the disease is very rare.
How is pycnodysostosis Treated?There is no treatment for the cause of PYCD, however injections of growth hormone have been shownto improve height. Plastic surgery can help correct deformities of the face and jaw.
Dental care will be necessary as people with PYCD are prone to cavities and may be missing teeth.Orthodontia is an option for improving the overall look to the teeth.
People with PYCD need to be careful with their bodies to avoid bone fractures. Exercise should belimited to low-impact activities such as swimming.
What is the Prognosis for Someone Withpycnodysostosis?The prognosis for a person with PYCD is generally good. He or she will be prone to fractures, but withcare, lifespan can be normal or near-normal.
Page 189 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Rhizomelic Chondrodysplasia PunctataType 1Gene: PEX7
Variant Accuracy PrecisionG217R 1.000 1.000L292X 1.000 1.000
Detection Rate Population57.10% African American57.10% Ashkenazi Jewish57.10% Eastern Asia57.10% Finland57.10% French Canadian or Cajun57.10% Hispanic57.10% Middle East57.10% Native American57.10% Northwestern Europe57.10% Oceania57.10% South Asia57.10% Southeast Asia57.10% Southern Europe
What is rhizomelic chondrodysplasia punctata type 1?Rhizomelic chondrodysplasia punctata type 1 (RCDP1) is an inherited disease that causes smallphysical size, certain characteristic bone problems, mental disability, and cataracts. Most children withthe classic form of RCDP1 do not live beyond the age of 10, and some will die in infancy. There isalso a mild form of the disease, but it is less common.
Classic Form
Children with the classic form of RCDP1 typically have shortened arm and leg bones. They are oftenborn smaller than average and fail to grow at the expected rate, leaving them much smaller than normalchildren. The cartilage in children with RCDP1 typically has round or oval areas of calcification.Affected children have stiff, painful joints which may lose the ability to bend normally. They will alsohave characteristic facial features.
Children with this disease are often severely mentally disabled and fail to develop skills beyond thelevel of a normal six month-old. The majority also develop seizures.
Page 190 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Other symptoms that may be seen include rough and scaly skin, a cleft palate, and malformations ofthe spinal column. These children usually develop cataracts early in life that obscure their vision. Mosthave recurrent lung infections which can be life-threatening.
Mild Form
In the mild form of the disease, mental and growth disability are less severe. Some have shortenedlimbs while others do not. All children with this form of the disease have areas of calcification in theircartilage and cataracts.
How Common is rhizomelic chondrodysplasia punctatatype 1?Fewer than 1 in 100,000 infants worldwide is affected by RCDP1. The disease affects children ofevery ethnicity, however one common mutation known as L292X is most common in Caucasians ofNorthern European descent.
How is rhizomelic chondrodysplasia punctata type 1Treated?There is no cure for RCDP1. Surgery to remove cataracts can restore some vision. Physical therapymay help preserve movement. Other bone surgery may also be helpful. Many children with the diseaserequire a feeding tube. Their lung function must be closely monitored to avoid infection and chokinghazards.
Those with milder forms of the disease may benefit from a specialized diet.
What is the Prognosis for Someone With rhizomelicchondrodysplasia punctata type 1?The prognosis for a child with the classic form of RCDP1 is poor. Many die in the first or second yearof life, and few survive beyond the age of 10. Breathing problems are often the cause of death.
Those with milder forms of the disease may live longer, however there have been relatively few knowncases with which to determine average longevity.
Page 191 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Salla DiseaseGene: SLC17A5
Variant Accuracy PrecisionLeu336fsX13 1.000 1.000R39C 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia
95.00% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Salla disease?Salla disease, also called free sialic acid storage disease, is an inherited condition causing a slow,progressive decline in motor and mental skills. Adults with the disease are profoundly disabled, butlive normal lifespans. Salla disease belongs to a group of diseases known as lysosomal storagedisorders.
Children with Salla disease appear normal at birth, but show poor muscle tone in the first year of life.Delays in their motor and mental skills become more obvious with age. They become spastic and willhave difficulty coordinating their voluntary movements. Most will be able to walk in adulthood,though some cannot.
Loss of intellect is progressive over time, beginning in the first or second year of life. Children withSalla disease typically have delayed language skills. By adulthood, all people with Salla disease areprofoundly mentally disabled, with IQs between 20 and 40. Most can speak some words in shortsentences.
In a more severe form of Salla disease, also called intermediate severe Salla disease, symptoms appearbetween the ages of 1 and 6 months. Infants have extremely poor muscle tone, growth delay, and mayhave seizures. Their loss of motor and mental functions is more rapid, and lifespan may be shortened.
Page 192 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Salla disease?Salla disease is rare except in Northern Finland, where 1 in 40 people are carriers. Only 30 cases ofintermediate severe Salla disease have been documented outside of Finland.
How is Salla disease Treated?There is no effective treatment for Salla disease other than to address symptoms as they arise. Specialeducation or physical, occupational, or speech therapy may be helpful.
What is the Prognosis for Someone With Salla disease?People with Salla disease have normal lifespans, but all will be profoundly disabled and will havedifficulty with movement. Most will be able to walk, but some will not.
Those with the severe form of Salla disease will have a reduced lifespan. The small number of casesknown worldwide make an exact prognosis difficult.
Page 193 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Segawa SyndromeGene: TH
Variant Accuracy PrecisionR233H 1.000 1.000
Detection Rate Population<10% African American<10% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Segawa syndrome?Segawa syndrome, also called dopa-responsive dystonia, is an inherited disease that can cause physicalrigidity and developmental delay. There are two forms: mild and severe.
In the mild form, symptoms typically begin in childhood. Children develop jerky movements thatquickly progress to physical rigidity. These children show spastic movements, and make very littlevoluntary movement. If untreated, children with Segawa syndrome may have expressionless faces,drooping eyelids, tongue tremors, and drooling problems. They will show both mental and physicaldevelopmental delays. Some children with Segawa syndrome show a “diurnal” pattern, meaning theirsymptoms are more or less severe on alternate days. With early treatment, children with Segawasyndrome can avoid many or all of the disease’s symptoms.
The severe form of the disease will appear in infancy, usually before six months of age. Affectedinfants have delayed motor skills, weakness in the chest and abdomen, rigidity in the arms and legs,and problems with movement. These children will eventually have learning disabilities, problems withspeech, and behavioral/psychological problems. In addition, some people with the disease haveproblems with their autonomic nervous system, which regulates unconscious functions such as bodytemperature regulation, digestion, blood sugar level, and blood pressure. Treatment of the severe formof the disease is often less successful.
Page 194 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Segawa syndrome is caused by a deficiency in an enzyme called tyrosine hydroxylase. Without it, theamino acid tyrosine cannot properly be converted to dopamine, a key neurotransmitter in the brain.Dopamine is important for many functions, including muscle control and cognition.
Note that there is another type of Segawa syndrome with a different genetic basis that is not addressedhere.
How Common is Segawa syndrome?The prevalence of Segawa syndrome is unknown, and only a small number of cases have beendiagnosed globally. Cases have been reported in Japan and in the Netherlands.
How is Segawa syndrome Treated?Individuals with the mild form of Segawa syndrome respond well to treatment with supplements of L-dopa and carbidopa. If taken before symptoms appear, the symptoms may be avoided completely. Ifsymptoms have already begun, children with the disease often respond extremely well to themedication, returning to normal very quickly. If the disease has gone untreated for some time, certainsymptoms may remain, including an irregular gait and other mild movement and speech difficulties.
Treatment with L-dopa and carbidopa supplements has been less beneficial for individuals with severeSegawa syndrome, but this treatment may improve motor skills over time.
If symptoms have gone untreated, physical, occupational, and/or speech therapists may prove helpful.
What is the Prognosis for Someone With Segawasyndrome?With early and consistent treatment, the prognosis for a person with mild Segawa syndrome is good.Many symptoms can be reversed with treatment. If treatment is not begun early and/or the course ofthe disease is severe, the person may be shorter than they would otherwise have been and may have anirregular walk and/or learning disabilities.
Page 195 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Short Chain Acyl-CoA DehydrogenaseDeficiencyGene: ACADS
Variant Accuracy PrecisionR107C 1.000 1.000G185S 1.000 1.000
Detection Rate Population<10% African American
65.00% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is short chain acyl-CoA dehydrogenase deficiency?Short-chain acyl-CoA dehydrogenase (SCAD) deficiency is an inherited disease in which the bodycannot turn certain fats (known as short-chain fatty acids) into energy due to a deficient enzyme. Itssymptoms can be triggered by illness or long periods without food.
Infants affected by the disease may display episodes of vomiting, low blood sugar, and fatigue. Theseepisodes can be fatal. Affected infants may have difficulty feeding and fail to grow at the expectedrate. Some show poor muscle tone, seizures, and small head size. If the disease is untreated, the childmay show developmental delays and permanent learning difficulties.
Some people with SCAD deficiency do not display any symptoms until adulthood. In these cases, themain symptom is chronic muscle weakness. Some may experience periods of pain, nausea, andshortness of breath. It is thought that many cases are so mild that they are never diagnosed.
SCAD deficiency belongs to a group of diseases known as fatty acid oxidation disorders.
Page 196 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is short chain acyl-CoA dehydrogenasedeficiency?SCAD deficiency affects 1 in every 40,000 to 100,000 newborns. Researchers have hypothesized thatthis disease may be more common because some people with the disease are asymptomatic or havemild symptoms.
How is short chain acyl-CoA dehydrogenase deficiencyTreated?It is critical that people with SCAD deficiency avoid going long periods of time without food. Infantsand children with SCAD deficiency may require feedings at regular intervals throughout the night. Acornstarch paste is often recommended to provide a sustained release of energy between meals. Foodshigh in carbohydrates and low in fat are also recommended. During times of illness, dietary rules mustbe very carefully followed. If the child cannot eat food for any reason, intravenous glucose must beadministered promptly.
Some physicians recommend carnitine supplements for people with SCAD deficiency.
What is the Prognosis for Someone With short chain acyl-CoA dehydrogenase deficiency?The prognosis for a person with SCAD deficiency varies widely and depends upon the severity of hisor her symptoms. In some cases, infants with the disease can die early in life. The prognosis for thosewho live into adolescence and adulthood and/or develop symptoms of muscle weakness later in life isnot known. Some people with the mutations that cause this disease do not develop symptoms, or havemild undiagnosed symptoms.
Page 197 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Sickle Cell DiseaseGene: HBB
Variant Accuracy PrecisionHb S 1.000 1.000Poly A: AATAAA->AATGAA 1.000 1.000Poly A: AATAAA->AATAAG 1.000 1.000W15X 1.000 1.000Pro5fs 1.000 1.000Gly16fs 1.000 1.000Glu6fs 0.992 1.000Lys8fs 1.000 1.000Phe71fs 1.000 1.000IVS-II-705 1.000 1.000IVS-II-844 1.000 1.000Gly24 T>A 1.000 1.000-30T>A 1.000 1.000CAP+1 A>C 0.997 1.000-87C>G 1.000 1.000Hb C 1.000 1.000Hb E 1.000 1.000Hb D-Punjab 1.000 1.000Hb O-Arab 1.000 1.000K17X 1.000 1.000Q39X 1.000 1.000619 bp deletion 1.000 1.000Phe41fs 1.000 1.000Ser9fs 1.000 1.000IVS-II-654 1.000 1.000IVS-II-745 1.000 1.000IVS-II-850 1.000 1.000IVS-I-6 1.000 1.000IVS-I-110 1.000 1.000IVS-I-5 1.000 1.000IVS-I-1(G>A) 1.000 1.000IVS-I-1(G>T) 1.000 1.000IVS-II-849(A>C) 1.000 1.000IVS-II-849(A>G) 1.000 1.000-88C>T 1.000 1.000-28A>G 1.000 1.000-29A>G 1.000 1.000
Page 198 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Detection Rate Population>99% African American>99% Ashkenazi Jewish>99% Eastern Asia>99% Finland>99% French Canadian or Cajun>99% Hispanic>99% Middle East>99% Native American>99% Northwestern Europe>99% Oceania>99% South Asia>99% Southeast Asia>99% Southern Europe
What is sickle cell disease?Sickle cell disease is an inherited disease that affects hemoglobin, the molecule in red blood cells thatcarries oxygen. Hemoglobin is made up of two different oxygen-carrying proteins, alpha and beta.People with sickle cell disease produce an abnormal type of beta protein. This results in red blood cellshaving a stiff crescent shape resembling a sickle. The sickled blood cells die prematurely, causing aperson to feel weak and tired, a condition known as anemia. These sickled cells also get stuck in smallblood vessels, blocking blood flow and causing serious medical complications such as blood-starvedorgans or tissue deterioration.
In a common form of this disease, called sickle cell anemia, the mutation that causes sickling (Hb S) ispresent in both beta protein genes. In other forms of sickle cell disease, the Hb S mutation is found inone beta protein gene and a different mutation (one that causes beta thalassemia) is found in the otherbeta protein gene. This means that a carrier of the Hb S mutation whose partner is a carrier of a betathalassemia mutation is at risk for having a child with sickle cell disease.
People with sickle cell anemia develop symptoms including anemia, repeated infections, shortness ofbreath, fatigue, jaundice, and bone pain starting in early childhood. The most recognizable symptom isepisodes of acute back, chest, or abdominal pain called "crises." These can last from hours to days.Crises are caused when the damaged blood cells break down, when the spleen enlarges to trap theblood cells, or when an infection makes the bone marrow stop producing red blood cells. Crises can berare or may occur multiple times a year. They can be severe enough to require hospitalization for paincontrol. Repeated crises can damage the organs and nervous system.
For those with other forms of sickle cell disease, the symptoms can vary from being as severe as sicklecell anemia to being mild.
People who are carriers of sickle cell disease are often said to have "sickle cell trait." Their hemoglobinmay be slightly abnormal, but typically they do not experience any symptoms of the disease.
Page 199 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is sickle cell disease?Sickle cell disease is common in people from Africa, the Mediterranean, the Arabian Peninsula, India,South America, and Central America. The large percentage of carriers in these regions is attributed tothe sickle cell mutation's protective effect against malaria.
Sickle cell anemia is very common in the African American population, where the disease affects anestimated 1 in 500 people and 8 to 10% are carriers. In people of Hispanic American background, thedisease affects an estimated 1 in 1,200 people.
How is sickle cell disease Treated?The symptoms of sickle cell disease can vary in severity, depending upon the mutations that a personcarries. Sickle cell anemia shows the most severe symptoms.
Sickle cell anemia can be cured with bone marrow transplants, but the procedure is extremely risky,both because the drugs needed to make the transplant possible are highly toxic and because it can bedifficult to find suitable donors.
For patients who are not candidates for bone marrow transplantation, sickle cell anemia requireslifelong care to manage and control symptoms and limit the frequency of crises. Fortunately, a betterunderstanding of how to manage the illness has extended patients' average lifespan by a decade ormore.
Preventing dehydration and avoiding infection can fend off crises and may prevent the sickling of redblood cells. People with sickle cell anemia, particularly children, should drink plenty of water, avoiddemanding physical activity and too much sun exposure, and get all appropriate vaccines andimmunizations.
Crisis episodes are treated with liquids to counter dehydration and medication to treat pain. Althoughmany crises respond to acetaminophen or ibuprofen, some will cause pain so severe they requirenarcotics. Other medication may help some patients whose crises include chest pain and difficultybreathing. Dietary supplements containing folic acid are important to help the body produce red bloodcells.
Other treatments that may be necessary as the disease progresses include dialysis or kidney transplant,gallbladder removal, spleen removal, eye surgery, hip replacement, and blood transfusion.
Page 200 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With sickle celldisease?People with sickle cell anemia generally die of organ failure or infection in their 40s or 50s. Frequentcrises and extreme fatigue can impair quality of life. People with other forms of sickle cell diseasehave varying degrees of symptoms.
Page 201 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Sjogren-Larsson SyndromeGene: ALDH3A2
Variant Accuracy PrecisionP315S 1.000 1.000
Detection Rate Population<10% African American
24.00% Ashkenazi Jewish<10% Eastern Asia
24.00% Finland24.00% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
24.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
24.00% Southern Europe
What is Sjogren-Larsson syndrome?Sjogren-Larsson syndrome (SLS) is an inherited condition with symptoms including dry, thickened,scaling skin (ichthyosis), spastic movement in the legs, and mental disability.
People with SLS cannot properly break down molecules called fatty aldehydes. The accumulation ofthese and related molecules results in the symptoms of SLS.
Children with SLS are often born several weeks prematurely and the majority either have ichthyosis atbirth or develop it in the first few months of life. Most of the remainder will develop this symptomwithin the first year of life. People with SLS will have thicker than normal skin that frequently itches.Scales on the skin can range in form from fine particles to large plate-like scales and they may be darkin color. Often the facial skin is unaffected.
Within the first two years, the nervous system often shows signs of the disease. These signs mayinclude developmental delay, speech abnormality, and seizures. Many people with the disease havemild to moderate mental disability. In terms of mental development, people with SLS do not worsenover time and can learn new skills.
Many people with SLS have spastic movement in their legs and the majority never learn to walk. Theymay also lose mobility in the joints of their lower body, contributing to motor problems. Spasticmovement can also affect the arms. Overall, people with SLS tend to be shorter than average.
Page 202 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Some people with SLS are extremely sensitive to bright light and many have glistening white dots inthe retina of the eye.
How Common is Sjogren-Larsson syndrome?The disease is most common in Sweden, where an estimated 0.4 out of 100,000 are affected by it. Innorthern Sweden, 1% of the population is a carrier of the disease.
Cases of SLS have been seen in people of all ethnicities. It is rare, though its exact frequency isunknown.
How is Sjogren-Larsson syndrome Treated?There is no treatment for the root cause of SLS, but its symptoms can be addressed.
Treatments for the skin problems caused by ichthyosis include daily baths, moisturizing creams, andcreams or lotions with active ingredients that slough off dead skin cells. Drugs called retinoids mayimprove skin condition, although they are not often used in children. Recent research has shown that adrug called zileuton can help reduce skin itching associated with SLS, but it is not yet FDA approvedfor this purpose. Some studies suggest that eating a diet with limited fats and taking medium-chaintriglyceride supplements can help ichthyosis, but the evidence so far has been mixed.
If a person with SLS has seizures, these usually can be controlled with anti-seizure medication.
Physical therapy may help build or regain motor skills including walking. Surgery may help reduce thespastic movements of the leg. Mechanical braces or other aids may be useful in helping people withSLS to walk.
What is the Prognosis for Someone With Sjogren-Larssonsyndrome?With good medical care, most people with SLS survive into adulthood. Their life expectancy may berelated to the degree of neurological symptoms.
Page 203 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Smith-Lemli-Opitz SyndromeGene: DHCR7
Variant Accuracy PrecisionL109P 1.000 1.000L157P 1.000 1.000R352Q 1.000 1.000R352W 1.000 1.000C380Y 1.000 1.000R404C 1.000 1.000IVS8-1G>C 1.000 1.000T93M 1.000 1.000W151X(c.452G>A) 1.000 1.000V326L 1.000 1.000W151X(c.453G>A) 1.000 1.000
Detection Rate Population59.26% African American59.26% Ashkenazi Jewish59.26% Eastern Asia59.26% Finland59.26% French Canadian or Cajun59.26% Hispanic59.26% Middle East59.26% Native American59.26% Northwestern Europe59.26% Oceania59.26% South Asia59.26% Southeast Asia59.26% Southern Europe
What is Smith-Lemli-Opitz syndrome?Smith-Lemli-Opitz syndrome, or SLO syndrome, is an inherited disorder in which the body's ability tomake cholesterol is impaired due to a deficient enzyme. Cholesterol is critical for the structure of cells,and is necessary for normal fetal development. It also plays an important role in the production ofhormones and digestive acids. In addition to low cholesterol levels, SLO syndrome also causes toxicbyproducts of cholesterol production to build up throughout the body, further disrupting growth anddevelopment.
In children with little or no ability to make cholesterol, symptoms are severe. These infants arecommonly born with an abnormally small head, cleft palate, and weak muscle tone. They often havedifficulty feeding because they lack the sucking reflex or have an abnormally small stomach that
Page 204 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

causes persistent vomiting. Some have extra fingers or toes as well as the typical fused second andthird toes on both feet. Male infants may have deformed or underdeveloped genitalia.
Infants with the severe form of SLO syndrome grow slowly and 90% have moderate to severe mentaldisability. Severely affected infants may also have heart defects and problems with their kidneys,causing death in the first months of life.
Some children are born with a milder form of the condition in which the body can produce somecholesterol. Symptoms may include developmental delays, feet with the second and third toes fusedtogether, slow growth, and short stature. These children generally learn to walk and talk and canacquire other skills, although they can rarely live independently as adults. Adults with the disease oftenshow aggressive behavior.
Symptoms of the disease can vary from person to person. Some affected people have only minorsymptoms of the condition.
How Common is Smith-Lemli-Opitz syndrome?Smith-Lemli-Opitz syndrome affects an estimated 1 in 20,000 to 60,000 people. This disease is morecommon in those of European ancestry, particularly those in Slovakia and the Czech Republic. It isvery rare among people of African and Asian descent.
How is Smith-Lemli-Opitz syndrome Treated?There is no cure for SLO syndrome, but its symptoms can be addressed. The primary treatment is tosupplement the person's diet with large amounts of dietary cholesterol, either in the form of purifiedcholesterol or in foods such as egg yolks and cream. This has been shown to improve symptoms. Earlyintervention and therapy helps with speech and physical disabilities. Medication may treat symptomssuch as vomiting, constipation, and gastroesophageal reflux. Surgery and orthotics can help musclespasms and improve mobility.
Because the condition can cause extreme sun sensitivity, people with SLO syndrome should alwayswear sunblock, sunglasses, and appropriate clothing when they go outdoors.
What is the Prognosis for Someone With Smith-Lemli-Opitz syndrome?Although serious internal malformations can lead to early death, with good nutrition and medical caremany people with SLO syndrome can have a normal lifespan. Mental disability typically preventspeople with this disease from living independently.
Page 205 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55
Spinal Muscular AtrophyGene: SMN1
Variant Accuracy PrecisionExon 7 deletion 1.000 1.000
Detection Rate Population71.10% African American90.20% Ashkenazi Jewish92.60% Eastern Asia95.00% Finland95.00% French Canadian or Cajun90.60% Hispanic95.00% Middle East95.00% Native American95.00% Northwestern Europe95.00% Oceania95.00% South Asia92.60% Southeast Asia95.00% Southern Europe
What is spinal muscular atrophy?Spinal muscular atrophy (SMA) is a disease in which certain nerves in the brain and spinal cord die,impairing the person’s ability to move. Called motor neurons, these nerves control our ability to sit up,crawl, and walk. In severe cases, a person will not be able to sit up independently and their breathingand swallowing may also be impaired. In the mildest cases, symptoms begin in adulthood and makeindependent movement such as walking more difficult, but still possible. There are five main subtypesof spinal muscular atrophy, each described below. It is not always possible to predict which type ofSMA a child could have based on the genetic mutation he or she inherits. This is true of the mutation(exon 7 deletion) for which Counsyl tests.
Type 0
Type 0 is the most severe form of SMA. Symptoms can often be seen in the later stages of pregnancyas the fetus is less active than expected. Once born, the infant will have little ability to move and maynot be able to breathe and swallow independently. Infants with type 0 SMA often die before the age of6 months.
Type I – Also Called Werdnig-Hoffmann Disease
Type I is another severe form of the disease. Symptoms develop within the first six months of life.Infants with SMA type I often have trouble breathing and swallowing. Their muscle tone and strength
Page 206 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

are extremely poor; they cannot sit up without support and will not meet any motor skills milestones.Their intellect, however, is normal. Most children with type I SMA will die before the age of two.
Type II – Also Called Dubowitz Disease
In children with type II SMA, muscle weakness becomes apparent between the ages of 6 and 12months. When placed in a sitting position, people with type II SMA can usually maintain the positionwithout support, however they often lose this ability by their mid-teens. People with SMA type IIcannot stand or walk without assistance. They have poor muscle tone and strength and their fingersusually tremble uncontrollably. Their intelligence is typically normal or above average.
Type III – Also Called Kugelberg-Welander Disease
Type III SMA is a milder form of the disease. Its symptoms begin sometime between the age of oneyear and early adulthood. As young children, they may fall repeatedly and have trouble walking downstairs. While their muscles are weaker than normal, people with type III SMA can usually stand andwalk without assistance, although they may lose this ability later in life. The legs are often moreseverely affected than the arms.
Type IV
Type IV is the mildest form of spinal muscular atrophy. With this form of the disease, muscleweakness does not begin until one’s 20s or 30s, or even later. This weakness is often mild to moderate,and the person can still walk and move independently. These individuals may experience mild tomoderate tremors and/or twitching. The disease typically does not diminish lifespan. With all types ofSMA, there can be difficulties in sleeping and gaining weight. Frequent pneumonia is common. Acurvature of the spine and stiff joints are also common. Women with milder forms the disease havebeen known to give birth to healthy children, although many of the pregnancies had complications.Thedisease is caused by a shortage in SMN protein, which helps preserve motor neurons. Without it, theneurons cannot pass messages from the brain to the muscles of the body.
How Common is spinal muscular atrophy?In the United States, 1 in every 6,000 to 10,000 people develop spinal muscular atrophy and 1 in 50 isa carrier of the disease. It has been found in people of every race, but is most common in Caucasians,of whom 1 in 35 is a carrier. Carrier rates for other populations include: Ashkenazi Jews (1 in 41 to62), Asians (1 in 53), African Americans (1 in 66), and Hispanics (1 in 117). Studies done in specificpopulations have found carrier rates of 1 in 50 in Germany, 1 in 57 in Italy, and 1 in 62 in China.
How is spinal muscular atrophy Treated?There is no cure for spinal muscular atrophy, however some of its symptoms can be addressed.
Page 207 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

For children with the more severe forms of spinal muscular atrophy, mechanical breathing aids mayprolong lifespan. In some cases, breathing is more difficult at night, leading to a lack of sleep. In thosecases, certain types of respiratory assistance may be helpful. If getting enough nutrition is an issue,some people with SMA have turned to feeding tubes.
Those with milder forms of the disease sometimes choose to have surgery to correct curvature of thespine (scoliosis) or joint problems. In forms of the diseases that are fatal in early childhood, thesesurgeries are often not done.
What is the Prognosis for Someone With spinal muscularatrophy?The prognosis for a person with SMA varies greatly depending on which type of the disease he or shehas.
Type 0
The disease is typically fatal between 2 and 6 months of age. These infants do not develop any motorskills expected of infants their age.
Type I
This type of SMA is usually fatal within two years. With mechanical breathing aids, children with typeI SMA may live longer. There are a few known cases of SMA type I in which the child survived toadolescence or adulthood.
Type II
With type II SMA, 75% of those affected live to the age of 25. They are often able to sit independentlywhen placed in a sitting position, but lose this ability by their mid-teens.
Type III
People with type III SMA may live a normal lifespan. Many learn to walk independently, though mostlose the ability to do so by their 30s or 40s.
Type IV
A normal lifespan is possible for people with type IV SMA. They do not develop symptoms until their20s or 30s and usually retain the ability to walk independently.
Page 208 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Sulfate Transporter-RelatedOsteochondrodysplasiaIncluding Achondrogenesis Type 1B, Diastrophic Dysplasia, and Recessive Multiple Epiphyseal Dysplasia
Gene: SLC26A2
Variant Accuracy PrecisionC653S 1.000 1.000R178X 1.000 1.000R279W 1.000 1.000V340del 1.000 1.000IVS1+2T>C 1.000 1.000
Detection Rate Population70.00% African American70.00% Ashkenazi Jewish70.00% Eastern Asia70.00% Finland70.00% French Canadian or Cajun70.00% Hispanic70.00% Middle East70.00% Native American70.00% Northwestern Europe70.00% Oceania70.00% South Asia70.00% Southeast Asia70.00% Southern Europe
What is sulfate transporter-relatedosteochondrodysplasia?Sulfate transporter-related osteochondrodysplasias are a group of inherited diseases caused bymutations in a gene important in cartilage and bone formation, called SLC26A2. These diseasesinclude: achondrogenesis type 1B, diastrophic dysplasia, and recessive multiple epiphyseal dysplasia.
Achondrogenesis type 1B
Achondrogenesis type 1B (ACG1B) is a severe skeletal disease that is fatal either before or shortlyafter birth.
Infants born with the disease have extremely short arms, legs, fingers, and toes. The fingers and toesmay be rotated inward. Infants with the disease also tend to have flat faces, protruding abdomens,narrow chests, and short necks that show thickening of the soft tissue. Many are born with hernias.
Page 209 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Fetuses with ACG1B are often in the breech position, “upside-down” with their feet toward the birthcanal. Mothers of fetuses with ACG1B are prone to certain other pregnancy complications.
Diastrophic dysplasia
Diastrophic dysplasia, also called diastrophic dwarfism, causes a person to be extremely small instature. It also causes joint pain, difficulty with movement, and bone and joint abnormalities.
People with diastrophic dysplasia have very short arms and legs, although their skulls are oftennormally sized. They are often born with bone deformities such as club foot, cleft palate, a curvedspine, and "hitchhiker thumbs" which are bent back. The outside of the ears are often swollen at birthand this can result in abnormal-looking ears later in life. People with the disease also tend to havesmall chests and protruding abdomens. The disease can cause breathing problems in infants,particularly due to the small size of the ribcage.
Those with the disease develop joint pain from an early age and have difficulty moving their joints.These symptoms worsen with age. Walking may become difficult for people with the disease. Adultheight of people with diastrophic dysplasia often ranges from 3.2 feet to 4.6 feet.
Diastrophic dysplasia does not typically affect intelligence or mental function.
Recessive multiple epiphyseal dysplasia
Recessive multiple epiphyseal dysplasia (rMED) causes bone deformities and joint pain. Unlike peoplewith related diseases, those with rMED typically reach normal height and live normal lifespans.
Half of people with rMED are born with an obvious bone abnormality such as cleft palate, club foot, oran inwardly-curved pinky finger. Some also have a mild curvature of the spine (scoliosis).
All people with the disease develop joint pain, often late in childhood. Pain is most common in the hipsand knees but can also occur in the wrists, fingers, and elsewhere.
How Common is sulfate transporter-relatedosteochondrodysplasia?ACG1B is very rare, and its frequency is unknown. One particular mutation that causes the disease ismost common in Finland, but other mutations are found globally.
Diastrophic dysplasia has been estimated to affect 1 in 100,000 people worldwide. It has been found inpeople of all ethnicities, but is most common in Finland.
Recessive multiple epiphyseal dysplasia is also rare, but researchers believe it may be more commonthan realized due to people with mild symptoms who go undiagnosed.
Page 210 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is sulfate transporter-related osteochondrodysplasiaTreated?There is no treatment for ACG1B. Infants with the disease can only be made as comfortable aspossible.
For people with diastrophic dysplasia, the goal of treatment is to improve and maintain mobility whilerelieving pain. This can be done with a combination of muscle exercises, surgery, and the use of plastercasts to hold childrens' joints in place. In particular, surgery can be used to correct club foot, to reducecompression of the spinal cord, or to correct knee joints. Surgery may need to be repeated as bonedeformities tend to re-form after surgery. It is important that people with diastrophic dysplasia do notbecome obese, as this puts harmful weight on their knee and ankle joints.
Recessive multiple epiphyseal dysplasia is usually treated through a combination of targeted musclestrengthening exercises and non-steroidal anti-inflammatory drugs (NSAIDs). People with the diseaseshould avoid sports and activities that stress their joints. Obesity too can put strain on the joints. Insome circumstances, surgery may be useful.
What is the Prognosis for Someone With sulfatetransporter-related osteochondrodysplasia?The prognosis for an infant with ACG1B is poor. They will die before or shortly after birth.
Infants with diastrophic dysplasia rarely face life-threatening breathing problems. Most people withdiastrophic dysplasia live into adulthood. All will face physical challenges with walking and othermovement, and may rely on various mechanical aids for mobility. They usually have normalintelligence and mental function.
People with recessive multiple epiphyseal dysplasia can live normal lifepans and can perform mostdaily activities, provided these don't stress the joints. Despite joint pain and some bone and jointabnormalities, people with rMED can live normal, healthy lives.
Page 211 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Tay-Sachs DiseaseGene: HEXA
Variant Accuracy Precision1278insTATC 1.000 1.000IVS12+1G>C 1.000 1.000IVS7+1G>A 1.000 1.000IVS9+1G>A 1.000 1.000
Detection Rate Population47.63% African American89.00% Ashkenazi Jewish47.63% Eastern Asia47.63% Finland47.63% French Canadian or Cajun47.63% Hispanic47.63% Middle East47.63% Native American47.63% Northwestern Europe47.63% Oceania47.63% South Asia47.63% Southeast Asia47.63% Southern Europe
What is Tay-Sachs disease?Tay-Sachs disease is a progressive condition that causes nerve cells in the brain and spinal cord to die,resulting in the gradual loss of movement and mental function. It is typically fatal early in childhood.
The symptoms of Tay-Sachs disease usually appear in infants between three and six months of age.Initially, infants lose the ability to turn over, sit, or crawl. They also become less attentive and developan exaggerated startle response to loud noise. As the disease progresses and nerve cells furtherdegenerate, infants with Tay-Sachs develop seizures, vision and hearing loss, mental disability, andeventually become paralyzed. Death usually occurs by the age of four.
Tay-Sachs disease, which is the most common and severe form of hexosaminidase A deficiency, iscaused by a deficiency in an enzyme called beta-hexosaminidase A. This enzyme helps break down aparticular fatty acid called GM2 ganglioside. Without adequate amounts of functional enzymes, GM2ganglioside will build up in nerve cells and cause them to die.
Page 212 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Tay-Sachs disease?Tay-Sachs disease is most common among specific ethnic populations, particularly Ashkenazi Jewsfrom Central and Eastern Europe, certain French-Canadian communities in Quebec, Amishpopulations in Pennsylvania, and Louisiana Cajuns. Tay-Sachs disease is found in people of allethnicities, though the risk outside of the ethnic groups mentioned above is much lower.
Roughly 1 in 30 Ashkenazi Jews is a carrier of Tay-Sachs, compared to 1 in 300 for the non-JewishCaucasian population. Since 1970, an organized campaign in the Jewish community to educatepotential parents about Tay-Sachs and test them for mutations causing this disease has dramaticallylowered the number of children affected by the disease. Because of these successful screeningprograms, today the majority of children born in the U.S. with Tay-Sachs disease do not have anAshkenazi Jewish background.
How is Tay-Sachs disease Treated?At this time there is no cure for Tay-Sachs disease, and treatment largely focuses around ensuring thechild’s proper nutrition and hydration, protecting his or her ability to breathe, managing any infections,and controlling seizures with medication.
What is the Prognosis for Someone With Tay-Sachsdisease?Even with the best care available, children affected by Tay-Sachs disease usually die by the age offour. They have worsening seizures and lapse into an unresponsive vegetative state.
Page 213 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

TPP1-Related Neuronal CeroidLipofuscinosisGene: TPP1
Variant Accuracy PrecisionG284V 1.000 1.000R208X 1.000 1.000IVS5-1G>C 1.000 1.000IVS5-1G>A 1.000 1.000
Detection Rate Population63.13% African American63.13% Ashkenazi Jewish63.13% Eastern Asia63.13% Finland63.13% French Canadian or Cajun63.13% Hispanic63.13% Middle East63.13% Native American63.13% Northwestern Europe63.13% Oceania63.13% South Asia63.13% Southeast Asia63.13% Southern Europe
What is TPP1-related neuronal ceroid lipofuscinosis?TPP1-related neuronal ceroid lipofuscinosis (NCL) is an inherited disease that causes degeneration ofthe brain leading to a progressive loss of mental and motor skills. It can also cause blindness andtypically leads to an early death. In the final stages of the disease, an affected person will be in avegetative state.
There are several forms of NCL, largely differentiated by the gene that carries the mutation and the ageat which symptoms begin. Mutations in the TPP1 gene typically result in the classic late infantile formor juvenile form of NCL.
Classic Late Infantile Form (LINCL)
The symptoms of classic LINCL typically begin between the ages of 2 and 4. Seizures are often thefirst sign, followed by a loss of the physical and mental milestones already achieved. Dementia soonfollows along with a loss of motor coordination. Children with classic LINCL become blind betweenthe ages of 4 and 6. They are often bedridden after the age of 6 and are unable to take care of
Page 214 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

themselves. Their life expectancy ranges from 6 to 40, with many succumbing to the disease by their20s.
Juvenile Form (JNCL)
The symptoms of JNCL, also called Batten disease, often begin between the ages of 4 and 10. Thesechildren rapidly lose their vision, becoming completely blind within two to four years. People withJNCL often develop periodic seizures between the ages of 5 and 18.
Between the ages of 8 and 14, mental functions typically decline. Children may have difficulty withspeech and show behavioral problems. Some people with JNCL also develop psychiatric problemsincluding disturbed thoughts, attention problems, and aggression. These problems can eventuallyprogress to dementia.
People with JNCL also show a decline in motor function and may have difficulty controlling their ownmovement.
How Common is TPP1-related neuronal ceroidlipofuscinosis?Approximately 1 in 25,000 people globally are affected by some form of NCL. These diseases aremost common in Scandinavian countries, but occur elsewhere as well. In the United States, it isestimated that 25,000 families are affected by some form of NCL.
Worldwide, 0.46 per 100,000 infants are born with TPP1-related NCL.
Mutations that cause TPP1-related NCL are more common in Iceland, Germany, and Finland than inother nations.
How is TPP1-related neuronal ceroid lipofuscinosisTreated?There is no treatment for the underlying cause of TPP1-related NCL. Treatments can only address thesymptoms as they arise. Various medications can be useful for treating seizures, poor muscle tone,sleep disorders, mood disorders, excessive drooling, and digestion. In some people, a feeding tube isalso helpful.
Page 215 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

What is the Prognosis for Someone With TPP1-relatedneuronal ceroid lipofuscinosis?The prognosis for people with TPP1-related NCL is generally poor. They will become blind and havesevere mental deterioration. They will enter a vegetative state in childhood and become totallydependent on others to care for them. Death can occur between the ages of 6 and 40.
Page 216 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Tyrosinemia Type IGene: FAH
Variant Accuracy PrecisionIVS8-1G>C 1.000 1.000P261L 1.000 1.000W262X 1.000 1.000E357X 1.000 1.000IVS12+5G>A 1.000 1.000
Detection Rate Population<10% African American
99.00% Ashkenazi Jewish<10% Eastern Asia
90.00% Finland>99% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American
50.00% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
50.00% Southern Europe
What is tyrosinemia type I?Tyrosinemia type I is an inherited metabolic disorder in which the body lacks an enzyme needed tobreak down the amino acid tyrosine, an important building block of proteins. The deficiency in thisenzyme, which is called fumarylacetoacetate hydrolase, leads to an accumulation of tyrosine andrelated substances in the body, which can damage tissues and organs.
There are several forms of tyrosinemia, but type I is the most severe. Symptoms will begin within thefirst few months of life. Early symptoms include diarrhea, vomiting, an enlarged liver, failure to growat a normal rate, yellowing of the skin and whites of the eyes (jaundice), a softening of the bones,irritability, and a boiled cabbage or rotten mushroom-like odor. Tyrosine will build up in the cornea,causing itchy, irritated eyes. The liver is progressively damaged, as are the kidneys and central nervoussystem. If left untreated, children with tyrosinemia type I may have episodes of abdominal pain, analtered mental state, pain or numbness in the extremities, and/or respiratory failure. A mechanicalventilator may be necessary for episodes of respiratory failure, which often last between one and sevendays.
Page 217 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

If not recognized and promptly treated, tyrosinemia type I is usually fatal before the age of 10. Death isoften due to liver or kidney failure, a neurological crisis, or hepatocellular carcinoma, a type of livercancer. Some children may die within weeks of experiencing the first symptoms. With treatment,however, 90% of people with the disease will live to adulthood and experience fairly normal lives.
How Common is tyrosinemia type I?Tyrosinemia type I affects 1 in 100,000 to 120,000 people worldwide. In the U.S., an estimated 1 in100 to 150 people are carriers of a genetic mutation that causes tyrosinemia type I. This disease ismore common in Norway and Finland, where it affects 1 in 60,000 births. It is also common inQuebec, Canada, where it affects 1 in 16,000 people. In the Saguenay-Lac-Saint-Jean region ofQuebec, the disease is especially common, affecting 1 in 1,846 people.
How is tyrosinemia type I Treated?The drug nitisinone (brand name: Orfadin) was FDA approved in 2002 to treat tyrosinemia type I. Itprevents an accumulation of specific metabolic compounds in people with the disease and is typicallytaken as soon as the disease is diagnosed.
The earlier the disease is recognized and treated, the less damage is done to the body and the better theprognosis. It is important that people with tyrosinemia type I manage their diets closely in a prescribedmanner to control intakes of tyrosine and another amino acid, phenylalanine.
Daily nitisinone intake and careful diet monitoring will be necessary throughout the life of someonewith tyrosinemia type I. Failure to comply with recommended treatments may result in the return ofsevere, potentially-fatal symptoms and damage to the body.
In severe cases where the affected person cannot take nitisinone or already has cancerous cells in theliver, liver transplantation is an option. The procedure does carry serious risks, however. Prior to thedevelopment of nitisinone, liver transplantation was the only treatment for tyrosinemia type I.
What is the Prognosis for Someone With tyrosinemia typeI?Without treatment, tyrosinemia type I is usually fatal by the age of 10 due to liver or kidney failure,neurological crisis, or liver cancer. However if promptly diagnosed and treated with nitisinone and amanaged diet, outcomes can be quite good with a survival rate greater than 90%. Children who receivethis treatment can grow to normal size and show improved liver and kidney function as well as morenormal bone structure.
Page 218 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Usher Syndrome Type 1FGene: PCDH15
Variant Accuracy PrecisionR245X 1.000 1.000
Detection Rate Population<10% African American
94.00% Ashkenazi Jewish<10% Eastern Asia<10% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Usher syndrome type 1F?Usher syndrome type 1F is an inherited disease that causes profound hearing loss from birth andimpairs vision beginning in adolescence.
Infants with Usher syndrome type 1F are profoundly deaf in both ears at birth. This hearing loss doesnot typically respond to hearing aids, though it may benefit from cochlear implants. Without earlyinterventions, the child may not develop speech abilities.
In adolescence, people with Usher syndrome type 1F develop retinitis pigmentosa, an eye diseasewhich causes night blindness and a gradual loss of peripheral vision. Eventually only the central visionremains, creating “tunnel vision.” This central vision too can be impaired and can lead to blindness in asmall number of people with the disease. In some cases, people with Usher syndrome type 1F developcataracts, which can further impair vision.
People with Usher syndrome type 1F also have severe balance problems and have trouble sensingchanges in speed or direction. They learn to sit and walk independently at a later age than otherchildren. Older children with the disease may appear clumsy due to their balance problems and mayhave difficulty with athletic activities.
The disease does not affect intelligence nor does it cause any other health problems.
Page 219 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Usher syndrome type 1F?Usher syndrome type 1—this includes subtypes 1A to 1G—affects 1 in 25,000 children. As many as 1in 70 U.S. adults may be carriers of some form of Usher syndrome. Approximately 11 to 19% ofpeople with Usher syndrome have type 1F.
Among children who are deaf or hard of hearing, 3 to 6% have some form of Usher syndrome—types1, 2, or 3. Among adults who are both deaf and blind, 50% have Usher syndrome.
Type 1F is more commonly found in Ashkenazi Jews. Between 0.7 and 2.5 in 100 Ashkenazi Jews arecarriers of a particular mutation that causes this disease, rating it one of the more commonly carrieddiseases among this population.
How is Usher syndrome type 1F Treated?There is no cure for Usher syndrome type 1F, however early treatment is important to give an affectedchild the best opportunity to develop communication skills. While a child is young, his or her brain ismost receptive to learning language, either spoken or signed. It is also important to take advantage ofthe time when the child’s vision is still normal.
People with Usher syndrome type 1F generally do not respond to hearing aids, however cochlearimplants may help regain some form of hearing. Sign language is a good option for communication.Specialists can introduce other tools and methods of instruction available to people with hearing loss. Itis often helpful if the whole family undergoes such instruction and as a family unit, helps the childadapt.
The child may also need low vision aids and specialized instruction on how to cope with his or herlimited vision. These children can be prone to accidental injury due to their vision loss and may needto devise systems to avoid such problems. For this reason, they may not want to participate in sports.Swimming is particularly difficult as people with Usher syndrome type 1F can become disoriented inwater.
What is the Prognosis for Someone With Usher syndrometype 1F?Usher syndrome type 1F causes severe hearing and vision impairment, however it does not affect one’slifespan or intelligence.
Page 220 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Usher Syndrome Type 3Gene: CLRN1
Variant Accuracy PrecisionN48K 1.000 1.000
Detection Rate Population<10% African American>99% Ashkenazi Jewish<10% Eastern Asia
0% Finland<10% French Canadian or Cajun<10% Hispanic<10% Middle East<10% Native American<10% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia<10% Southern Europe
What is Usher syndrome type 3?Usher syndrome type 3 is an inherited disease that causes progressive hearing loss and visionimpairment. The rate at which hearing and vision decline varies greatly from person to person, evenamong those in the same family. In some people, the hearing and/or vision loss can be profound, whilein others it can be milder.
People with Usher syndrome type 3 are born with normal hearing and most commonly develop hearingloss in their teenage years, requiring hearing aids by mid- to late-adulthood. By middle age, they areoften completely deaf.
Usher syndrome type 3 causes an eye disease known as retinitis pigmentosa. Often arising duringpuberty, this causes night blindness that progresses to blind spots in the late teens or early adult years.Peripheral (side) vision is often the first to be reduced. Often by mid-life, the person is legally blind.
Unlike other forms of Usher syndrome, type 3 does not usually cause major problems with balance.Some problems may arise later in life, however.
The disease does not affect intelligence nor does it cause any other health problems.
Page 221 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How Common is Usher syndrome type 3?Usher syndrome type 3 is rare, making up just 2% of all cases of Usher syndrome. Type 3 is morecommon in Finland and among Ashkenazi Jews. One study showed that in the New York City area,0.7% of Ashkenazi Jews are carriers of a particular mutation, which would mean that 1.2 in 100,000Ashkenazi Jewish children would be affected.
How is Usher syndrome type 3 Treated?There is no cure for Usher syndrome, however there are ways to negotiate the vision and hearing loss itcauses.
People with the disease will learn to speak normally before their hearing declines. They can explore arange of options including cochlear implants, hearing aids, or sign language.
A person with Usher syndrome will eventually require low vision aids and specialized instruction onhow to cope with their limited vision. They can be prone to accidental injury due to their vision lossand may need to devise systems to avoid such problems.
Specialists in both hearing loss and vision loss can guide people to the best options to fit their needs.
What is the Prognosis for Someone With Usher syndrometype 3?Usher syndrome type 3 will cause severe hearing and vision impairment by mid-life, however it doesnot affect one’s lifespan or intelligence.
Page 222 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Wilson DiseaseGene: ATP7B
Variant Accuracy Precision1340del4 1.000 1.000R778G 1.000 1.000W779X 1.000 1.0002337delC 1.000 1.000H1069Q 1.000 1.000
Detection Rate Population<10% African American
42.30% Ashkenazi Jewish<10% Eastern Asia
42.30% Finland42.30% French Canadian or Cajun
<10% Hispanic<10% Middle East<10% Native American
42.30% Northwestern Europe<10% Oceania<10% South Asia<10% Southeast Asia
42.30% Southern Europe
What is Wilson disease?Wilson disease is an inherited disease that causes the body to retain too much copper. Copper depositsin the liver, brain, kidneys, and eyes eventually cause tissue damage and scarring that makes theaffected organs stop working properly. If not diagnosed and treated early, the condition causes organfailure and death.
Symptoms typically first appear in childhood or early adolescence, but they can appear as early as age3 and as late as age 70. The most common symptom is liver disease, which first appears as fatigue,abdominal pain, or jaundice. In some cases, it progresses quickly to liver or kidney failure, and willrequire a liver transplant.
Symptoms can also include neurological problems such as tremors, poor coordination, loss of finemotor skills, problems walking, muscle rigidity in the body or face, or difficulty swallowing. Somepeople with the condition also develop psychiatric problems including depression, poor impulsecontrol, phobias, aggression, and compulsive behavior. Wilson disease may also interfere with memoryand attention span.
Page 223 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

Copper deposits also accumulate in the eyes, creating characteristic brown circles around the coloredpart of the eye. These circles do not interfere with vision.
Even with ongoing treatment to remove excess copper from the body, people with Wilson diseasesometimes develop arthritis, heart problems, and endocrine disorders caused by copper accumulation.
How Common is Wilson disease?Worldwide, approximately 1 in 30,000 people have Wilson disease. It is most common in China,Japan, and Sardinia, where it may affect as many as 1 in 10,000 people.
How is Wilson disease Treated?Wilson disease requires lifelong, regular treatment to remove copper from the body. Most people withthe condition take a medication called penicillamine (brand name: Cuprimine or Depen) several timesa day by mouth, combined with vitamin B6. This traps and removes copper from the body through theurine. However, some people react to penicillamine with fever, rash, and other serious complications.These people may be treated with other oral medications such as trientine or high-dose zinc. Forindividuals who do not respond to medication or have severe side effects, liver transplant is a finaltreatment option.
With careful treatment prior to the first symptom's appearance, most symptoms can be prevented. Iftreatment begins after symptoms appear, these symptoms can often show marked improvement.Stopping treatment, however, will cause health problems to return.
People with Wilson disease should not use copper cooking utensils. They should avoid foods high incopper, such as liver, chocolate, mushrooms, nuts, and shellfish. If they live in an area with copperwater pipes, they should drink distilled water.
What is the Prognosis for Someone With Wilson disease?With proper treatment, Wilson disease can be managed for many years after diagnosis. Its effect onlifespan is unclear.
Page 224 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

X-Linked Juvenile RetinoschisisGene: RS1
Variant Accuracy PrecisionE72K 1.000 1.000G74V 1.000 1.000G109R 1.000 1.000
Detection Rate Population14.39% African American14.39% Ashkenazi Jewish14.39% Eastern Asia95.00% Finland14.39% French Canadian or Cajun14.39% Hispanic14.39% Middle East14.39% Native American14.39% Northwestern Europe14.39% Oceania14.39% South Asia14.39% Southeast Asia14.39% Southern Europe
What is X-linked juvenile retinoschisis?X-linked juvenile retinoschisis is an inherited eye disorder that makes the inner layer of the retina splitin a spoke-wheeled pattern. It usually occurs in both eyes at once. This disease affects more males thanfemales.
Boys with the condition may have symptoms in early childhood, but are often not diagnosed until theyhave their first visual screening before starting school. They usually have vision of 20/60 to 20/120,with tiny lesions, splits, or cysts visible on their retinas. Fewer than 10% of people with the conditionexperience full retinal detachment or bleeding inside the eye, both of which can cause blindness.
As boys with X-linked juvenile retinoschisis go through their teens, their vision may slowly worsen,but it typically stabilizes in their twenties. When they reach their forties and fifities, their vision maystart to deteriorate again. Many people with the condition eventually have vision of 20/200 or worse,making them legally blind.
How Common is X-linked juvenile retinoschisis?X-linked juvenile retinoschisis is estimated to affect 1 in every 5,000 to 25,000 people. Due to itspattern of inheritance, the disease primarily affects males.
Page 225 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55

How is X-linked juvenile retinoschisis Treated?In general, treatment focuses on monitoring the progress of the disease and helping affectedindividuals learn to cope with poor vision. For example, children can use large-text books and high-contrast reading materials. Adults can purchase special magnifiers, clocks, and adaptive software tohelp them at home and at work.
Because X-linked juvenile retinoschisis affects only the inner layers of the retina, surgery is rarelyeffective in treating the disease. However surgery may help with complete retinal detachment.
Low-vision specialists such as optometrists can help both children and adults make the most of thevision they have. Some people with the condition can get restricted driver's licenses if they wearspecial telescopic lenses behind the wheel. (Please note that these lenses are not legal in all states.)
Children under the age of 10 should see a pediatric ophthalmologist or retina surgeon every year. Olderchildren and adults need less frequent monitoring.
People with X-linked juvenile retinoschisis should avoid high contact sports and other activities thatmight cause a hard blow to the head. This minimizes the risk of retinal detachment or bleeding in theeye.
What is the Prognosis for Someone With X-linked juvenileretinoschisis?X-linked juvenile retinoschisis does not affect lifespan, but does cause progressive vision problemswhich can result in legal blindness after middle age.
Page 226 of 226Counsyl Universal Genetic Test - Disease Book v1.0.55