1901 master - inis.iaea.org
TRANSCRIPT

"1901
MASTERA RE-DETERMINATION AND RE-ASSESSMENT OF THE
THERMODYNAMICS OF SUBLIMATION OF URANIUM DIOXIDE
by
R, J, Ackermann and E. G. Rauh
any winaniy, expittl <ir implied.
or uwlulneu ut tny minin\»\inn. •ppiuiui, pto<\mt »i
Prepared for
International Symposium on
The Thermodynamics of Sublimation of Uranium Dioxide
Julich, Germany
January 29 - February 2, 1979
UofC-AUA-USDOEARGONNE NATIONAL LABORATORY, ARGONNE, ILLINOIS
Operated under Contract W-31-109-Eng-38 for the
U. S. DEPARTMENT OF ENERGY

1. Introduction
A reliable equation of state for U0? and other nuclear fuel
materials up to 6000 K is needed for the safety analysis of
hypothetical core-disruptive accidents in fast-breeder reactors.
The vapor pressure of the oxide fuel is an important parameter
in the energy release mechanism and requires either a long extra-
polation of the lower temperature data (T < 3000 K) or new
methods of measurement at the temperatures of interest. Both
options have certain limitations peculiar to the chemical
thermodynamic behavior of all materials at high temperatures.
The vapor pressure and enthalpy of sublimation of UO~ have
been reported by many investigators using the Knudsen (mass
effusion), Langmiur (surface evaporation), mass spectrometric,
and transpiration methods. The results of the most extensive
measurements to date are given in Table 1. The results of
Ackermann et at. [1] have been separated traditionally into low
and high temperature regions as a result of the observed
positive curvature in the plot of log p against 1/T. It is
necessary to re-examine these data in the light of new measurements
and to re-interpret the high and low extremities of the original
curve. Hence, in Table 1 are given the thermodynamic quantities
derived from the most reliable central region (1758-2378 K) of
the temperature range that is defined by no net statistical

deviation from linearity of log p vs 1/T. From Table 1 one
concludes that the variety of methods of measurement yield at
2150 K, a temperature common to nearely all of the studies,
values of the vapor pressure which agree within a factor of
approximately two except that calculated from the anomalously
high results of Gross [5]. Only the mass-spectrometric
results of Pattoret et al. [8] correspond explicitly to the
gaseous molecule, UO?(g); all of the other measurements
involve the total uranium transport as UO2(g) and losser
amounts of UO(g) and UO^(g). At temperatures near 2150 K,
the combined partial pressures of the latter two are relatively
small 18,10] so that p - .94 Pfcot- Hence, the respectively
derived enthalpies and entropies of sublimation should closely
approximate that of UO2(g) if the temperature range is centered
around this temperature.
A further inspection of Table I indicates a spread of
values for AH ranging from 140 to 152 kcal/mole. This variation
adds considerably to the uncertainty resulting from an extra-
polation of the vapor pressure to much higher temperatures.
Furthermore, the very recent measurements of the vaporization
rates of U0?(£) above 4000 K by laser irradiation and momentum
detection or mass transfer methods [11-13] correspond to total
vapor pressures of many atmospheres that appear to be significantly

4
higher than the values for UO-(g) calculated from many of the
results given in Table 1. The molecularity of vaporization
at these high temperatures has not been identified and therefore,
it will be elucidated from a more thorough assessment of the
results given in Table 1 supplemented by the present study.
Several of the previous measurements of the vapor pressure
of U02 cited in Table 1 (Ackermann et al. [1] and Okse [4]),
were carried out by extending the effusion method to pressures
approaching 10 atm. The upper pressure limit of the effusion
method must be more carefully examined in the light of some
recent studies [14-16], which clearly show a breakdown of
-4effusive flow for pressures exceeding 10 atm.
It is, therefore, the purpose of this study to improve the
reliability of the thermodynamic data for the vaporization of
uranium dioxide by 1) presenting precise mass spectrometric
measurements of the enthalpy of sublimation to UO-(g) over a
wide (^ 700°) temperature range, 2) calculating the thermodynamic
functions for UO_(s and g) from the critical assessment of
presently available data, and, 3) analyzing the departure from
molecular flow and the failure of the Knudsen effusion method
at vapor pressures of uranium dioxide in excess of approximately
10~4 atm.

2. Experimental methods and results
Seven series of measurements of the temperature dependence
of the partial pressure of UO_(g) over approximately stoichiometric
UO2(s) were made mass-spectrometrically by observing the U02 ion
current as a function of the effusion cell temperature. The
observations were obtained with a Bendix model 12-101 time-of-
flight mass spectrometer equipped with a tungsten effusion cell
heated by electron bombardment. Details of the cell assembly [17],
power regulator [18] and the pyrometric measurement of temperature
[19] have been reported elsewhere. Three different samples of UOp
(200-350 mg) were used and the overall instrumental sensitivity
was changed from one series to another in an attempt
to identify systematic instrumental errors that might involve a
non-linear response. The possibility of error due to temperature
gradients was investigated by altering the position of the cell
with respect to the filament. When the cell was raised sufficiently,
enough of a gradient could be introduced so that a deposit formed
within the orifice; at the other extreme the temperature measured
through the orifice began to appear "non-black". Within these
limits the position of the cell did not effect the reproducibility
of the results within the experimental uncertainties. The
possibility of temperature errors at the extremes of the pyrometer

scales was minimized by supporting readings through a calibrated
neutral density filter which shifted the readings down scale.
The linearity of the ion-detection system was established by
measurements of the neon isotopes at varying gas pressures.
Each of the seven series of measurements consisted of 17-30
random observations of the UO2 ion current-temperature relationship
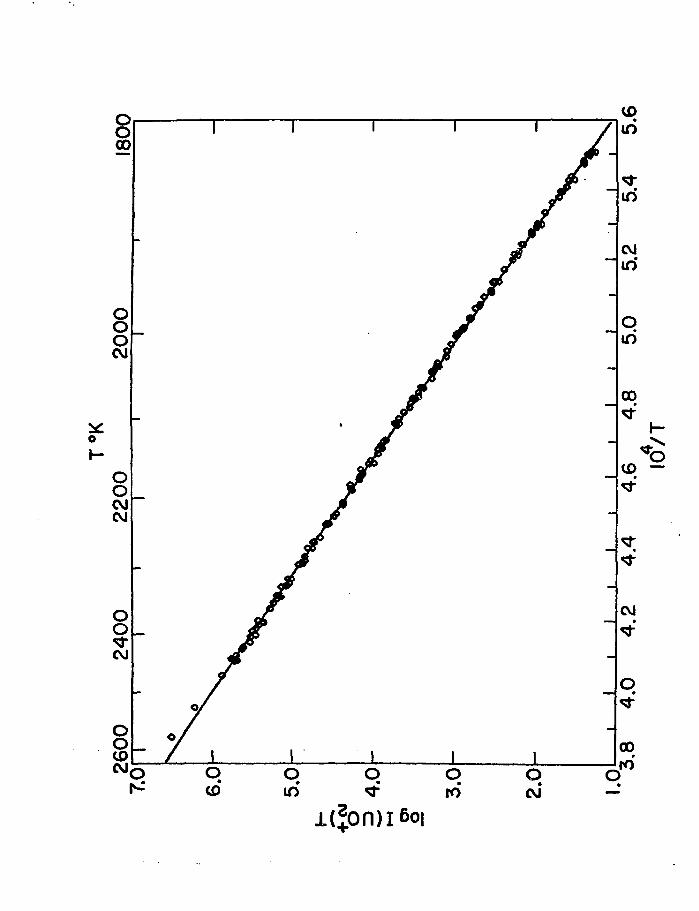
over the range 1813-2463 K. All the data normalized to ci common
sensitivity at 2150 K are plotted in fig. 1. The maximum useful
temperature was limited by tha high rate of loss of sample and
the rapid formation of deposits in the collimating slits as well
as by the possibility of exceeding molecular flow where ion
currents are not simply proportional to partial pressures. The
combined results of all runs (140 data points) were expressed
as log I(UO2)T vs 1/T and treated by the linear least squares
method. The normalization of all runs to an arbitrary ion
current at 2150 K yields the linear equation, log I(UO2)T =
(18.388 + 0.026) - (30927 + 60)/T from which the average enthalpy
of sublimation is 141.5 +0.3 kcal mol" , a value in excellent
agreement with that, 141.2 + 1.1 kcal mol" , reported by Pattoret
et at. [8].
A careful examination of the residuals shown in the upper
plot of fig. 2 indicates a curvature in the data beyond the
experimental uncertainties and a lack of a good statistical fit
of a simple linear representation even though the standard deviations

in the slope and intercept are small. The results of each series
were then fitted to a three parameter least squares equation
of the form, log I(UO2)T = A + ^ + C £n T, and normalized to
a common sensitivity at 2150 K. The residual pattern resulting
from this treatment is shown by the lower plot in fig. 2. The
plots of the residuals clearly demonstrate that a linear least
squares representation of the data is not quite adequate and
shows a trend with temperature whereas the three parameter
equation removes the residual curvature and shows no trend with
temperature. All of the experimental precautions and variations
in the measurements of ion currents and temperatures did not
remove this curvature or reveal any readily identifiable systematic
errors. The solid line, fig. 1, through the results will
be identified and discussed in much greater detail later.
Attempts were made to extend the temperature range beyond 2460 K
into the region of non-effusive flow, but the measurements were
severely limited by the rapid loss of sample. However, at the
end of one of the series
range 2470-2580 K which showed increased U02 ion intensities
and a definite reversal of the downward curvature. These are
shown in fig. 1 at the higher temperature end of the curve and
will be discussed in terms of the departure from molecular flow
that follows.

3. Discussion
The constant C in three-parameter equation that has been used
to fit and to normalize the data shown in fig. 1 is equal to the
average value of the heat capacity change divided by the gas
constant, i.<>., (AC~ /R) . Hence, values of AC"n can be obtained
from each individual series and from the combined normalized
data; these values vary from extremes of -2.C. to -20 cal mol~
deg . This variation is too large to obtain a reliable average
value and nost certainly precludes the functional derivation
of the nature of the dependence of "AC upon temperature. Therefore,P
the experimental data alone are inadequate as a result of the gentle-
ness of the curvature compared with the existing random as well as
unknown systematic errors. However, the results strongly indicate
that AC cannot be regarded as zero but must be quite negative.
A small but insignificant systematic error is present at
the higher temperatures for the data plotted in fig. 1. The
UO2(s) phase does not remain exactly stoichiometric over the
large temperature range of this study. In fact, the congruently-
vaporizing-composition departs from the composition UO- »n
above 2000 K and becomes progressively substoichiometric reaching
a composition UO, o o near 2450 K [20j . The extend of this
departure reduces the partial pressure of UO-(g) by less than
1% up to 2450 K and, hence, is considered insignificant.

In order to represent the vapor pressure of UO2(g) sufficiently
accurately within the present temperature range and to permit
extrapolations much beyond this range of measurements, a knowledge
of the variation of AC with temperature is needed to complement
the present results.
Uranium dioxide exists as a crystalline solid with the
fluorite structure at all temperatures below the melting point.
Materials having this structure are known to possess excess
contributions to the enthalpy and heat capacity at high temperatures
due to the formation principally of Frenkel defects in the anion
sublattice [21,22]. This behavior is a cooperative process
resembling fusion and is often described as pre-melting of the
anion sub-lattice. Bredig [23] has analyzed and discussed the
excess enthalpy in UO2(s) and related its ? ̂ilarity with that
in other fluorite-structured materials and has concluded that
a lambda-type transition indeed may occur near 2670 K. Rand
et at. [24] have separated the five sets of experimental
measurements of enthalpy (675-3112 K) and the two sets of heat
capacity (298-1006 K) into two regions, below and above 2670 K,
and have shown that Bredig's astute prediction is borne out
statistically. As a numerical convenience only, the lambda
transiti near 2670 K is taken as first-order and amounts to
an enthalpy change of 436 cal/mol. The thermodynamic functions
derived by Rand et al. [24] are given in Table 2.

10
The heat capacity and other thermodynamic properties of UO^
have not been measured but can be calculated from known molecular
parameters and some numerically consistent estimates of the
electronic contribution to the partition function that can be
closely fitted to the measured vapor pressure and enthalpy of
sublimation over the large temperature range of this study.
The need to know or to estimate reliably the low-lying electronic
states and degeneracies in gaseous molecules is essential to any
third-law treatment of the data that can be used for extrapolative
purposes. At the present time, however, there exists no reliable
theoretical basis for predicting the term values and degeneracies
of electronic states particularly in a many-electron molecule
such as UO^(g). Brewer and co-workers [25,26] have attempted to
approximate the electronic contribution in a gaseous molecule
with that for the gaseous charged atom, e.g., a gaseous diatomic
oxide, MO(g), is taken the same as the electronic partition
+2function for M (g) or its isoelectronic equivalent. These
approximations in general are too large by several cal mol deg
in the entropy and free energy function in cases for which the
electronic states in both the molecule and gaseous ion are
known [27,28],
At the mid-temperature (2150 K) of the present study the
total vapor pressure of all uranium-bearing species from the
experimental measurements given in Table 1 is 1.36 x 10 atm.

11
The partial pressure of UO2(g) is about 94 ± 3% this values based
on the mass spectrometric measurements of Ackermann et al. [29]
and Pattoret et al. [8]. If this factor is applied to the total
pressure measurements (both effusion and transpiration), an
"international average", PT = 1.29 x 10~ atm,is obtained for
the partial pressure of UO2(g) in equilibrium with virtually
stoichiometric UO~(s) at 2150 K. The average enthalpy of
sublimation of UO2(g) at 2150 K measured mass spectrometrically
in the present study is 141.5 kcal mol which agrees closely
with that (141.2) reported by Pattoret et al. From these values
of the pressure and its temperature dependence an entropy of
sublimation of 38.9 cal mol" deg" at 2150 K is obtained. The
absolute entropy of UO2(s) at 2150 K is 57.0 cal mol" deg" ;
hence, the absolute entropy of UO-(g) is 95.9 cal mol deg at
2150 K. It is possible to account for 91.5 cal mol deg
calculated from the known molecular parameters of UO2(g). The
molecule is known to be linear in its ground state from matrix-
isolation spectroscopy from which the measured stretching
frequencies, v. = 765.4 cm and v, = 776.1 cm , were determined.
A doubly degenerate bending frequency, v2 = 190 cm" , calculated
from the valence force model applied to the uranyl ion, UO? ,
is consistent with the stretching frequencies. A previously

12
observed absorption band at 81 cm was tentatively assigned to
the bending mode in UO2(g) but has not been confirmed and appearso
to be abnormally small. A U-0 distance of 1.79 A has been used
in these calculations. The breakdown of the Born-Oppenheimer
approximation at high temperatures produces numerically significant
anharmonocity corrections which are included in these calculations.
Hence, the contribution to the entropy by unknown electronic
states in UO-fg) may be nominally as large as 4.4 cal mol" deg~ .
The spectra for u (g) are not known, but those for isoelectronic+2 +3
Th (g) and Pa (g) [30] give rise to electronic entropies of 7.1
and 6.2 cal mol deg , respectively, which clearly are too large.
Therefore, we have constructed somewhat arbitrarily an electronic
partition function for UO2(g) using as a guide that for ThO(g)
which is the only gaseous actinide oxide that has been so
characterized. Both molecules have two unbonded electrons but
any further similarity is lacking. However, the thermodynamic
functions can be constructed in a consistent fashion from these
electronic estimates and the known parameters and they are given
in Table 2. A plot of the heat capacities of the gas and solid
as well as AC is shown in fig. 3. The resultant AC is seen toP P
be strongly temperature-dependent and can be adequately approximated
by a parabolic dependence on temperature up to the transition
temperature, 2670 K.

13
An equation of the form:
2
log p U Q = A + BT + CT + D/T + E Jin T (1)
which incorporates the parabolic dependence of AC on temperature,
AC = 2.30>rf2BT + 6CT2 + E], (2)
has been fitted to the data of the present study. The constants
B, C, and E were fixed from the dependence of AC on temperatureP
seen in fig. 3 and the remaining constants A and D were obtained
by least-squares treatment of all the ion-current data shown in
fig. 1 normalized to the value of pn_ = 1.29 x 10~ atm at
2150 K. The values of the constants are as follows:A ~ 67.531 (p/atm)
B = 4.382 x 10"3
C = -4.411 x 10"7
D = -3.7094 x 104
E = -8.282
Equation (1) with these constants gives rise to the curve drawn
through the data shown in fig. 1. The enthalpy of sublimation
of UO2(g) obtained by differentiating eq. (1) with respect to
1/T varies from 143.0 kcal mol~ at 1800 K to 138.5 kcal mol~

14
at 2400 K. The change of the enthalpy of sublimation with
temperature is dominated by the excess heat capacity of the
solid phase as seen in fig. 3. The uncertainties in the estimates
of the bending vibrational constant and the electronic states in
UO_(g) have only a very minor influence on the change in ACf and
hence AH° with temperature. A third-law treatment of the data
according to the equation, hU° = -4.576T log p - A(G£-H°/T), and
the necessary quantities from eq. (1) and Table 2 yields a virtually
constant value, of AH2 = 147.5 kcal mol having an insignificant
trend of only 0.2 kcal mol across the temperature range,
1800-24 00 K. A plot of the residuals resulting from the least
squares treatment by means of eq. (1) of all the data shown in
fig. 1 yields a distribution pattern that is virtually identical
to that shown in the lower plot of fig. 2, and consequently,
demonstrates the absence of any significant systematic errors.
The partial pressure of UO~(g) above UO2(s) calculated from
eq. (1) is plotted against reciprocal temperature in fig. 4 and
is compared with the total pressure of uranium-bearing species
reported in some of the earlier studies. The results of
Ackermann et al. [1] and Tetenbaum and Hunt [9] are virtually
indistinguishable up to 2450 K. Above this temperature the
results of Ohse [4] are in close agreement with the higher
temperature mee jurements of Ackermann et al. It is immediately

15
clear that these latter two results are substantially higher than
the curve for UO2(g) that has been fixed at 2150 K as described
above. All previous mass spectrometric studies agree that UO2(g)
is the major species vaporizing from congrently vaporizing UO2(s);
the only other observed molecules of any importance, UO(g) and
UO3(g), occur at relatively smaller concentrations. Therefore,
the ostensibly higher pressures of Ackermann et at. and Ohse
above 24 50 K cannot be explained by unobserved gaseous species
of sufficiently large partial pressures or by any reasonable
variation of the value of AC for the vaporization of UO-tg)
that results from an arbitrary adjustment of C (U02 ).
The findings of Carlson et at. [14] describe quantitatively
the increased mass effusion rate of saturated mercury vapor that
occurs from a thin-edged orifice in the transition from molecular
to hydrodynamic flow. The departure from molecular (effusive)
flow begins when the mean-free-path in the vapor becomes
approximately equal to the diameter of the orifice. This condition
-4will prevail when the saturated vapor pressure exceeds ^ 10 atm.
At higher temperatures, and hence, for larger vapor pressures,
the mass effusion rate increased to a limiting hydrodynamic value
near 1.6 that expected for molecular flow. These results, shown
in fig. 5, show a marked increase in the ratio of the pressure

16
p calculated from the measured rate of mass transport of mercury
vapor via the effusion equation, p = m^ Jj.RT' , to the saturated
vapor pressure p plotted against log p . Similarly, the results
of Ackermann et al. and Ohse for UO- show a marked increase for
the same condition. The saturated vapor pressure of UO,(g), p ,
has been calculated from eq. (1). Inspection of the various
results in fig. 5 rather strongly indicates that the effusion
-4method cannot be extended to pressures exceeding 10 atm for
conventional size orifices, 0.05-0.1 cm diameter, and a
corresponding mean-free-path of comparable magnitude. The
deviation from molecular flow progressively increases with increasing
pressures shown in fig. 5 and hence, as seen in fig. 4, causes
the apparent enthalpy of sublimation to increase. Schulz and
Searcy [15] have also shown in a convincing manner that molecular-
flow conditions break down for CaF_(g) when the mean-free path
approaches the orifice diameter. However, Ewing and Stern [16],
in a study of the vaporization of alkali halides have proposed
the unsuspected result that the transition from molecular to
hydrodynamic flow depends on the mean-free-path and is independent
of the diameter of the orifice. Although there still exists some
lack of agreement among these investigations in the identification
of the parameters explicitly associated with the break-down of

17
-4molecular flow, it is clear that such occurs near 10 atm and
has undoubtedly contributed an appreciable systmatic error in
the vapor pressure of UO2 measured at the higher temperatures.
Therefore, the values thereof derived from the high temperature
data of Ackermann et at. [1] and those of Ohse [8] exceeding
150 kcal mol are not valid quantities and should not be used
to extrapolate the vapor pressure to higher temperatures [13,32]
even for comparative purposes.
The reasons for the breakdown of the effusion method at
higher pressures do not apply to the transpiration technique
employed by Tetenbaum and Hunt [9]. The small discrepancy
between the transpiration results and those of p u 0 in the present
study is explainable in terms of the concentrations of UO(g) and
UO., (g) in the vapor which increase relative to that of UO-(g)
with increasing temperature. Also seen in fig. 4 as ©-points
are the three highest temperature points of the present study
shown previously in fig. 1. These points very likely indicate
the departure from molecular flow for U02(g) observed mass
spectrometrically. The B-points are the effusion results
reported earlier by Cater [31].
3/24/78 cac

Table 1
Thermodynamics of Sublimation of Uranium Dioxide
Investigators
Ackermann et at.(1956) [1]
Ivanov et at.(1962) [2]
Voronov et at.(1962) [3]
Ohse(1966) [4]
Gross(1966) [5]
Alexander et at.(1967)** [6]
Gorban et at.(1967) [7]
Pattoret et at.(1968) [8]
Tetenbaum andHunt (1970) [9]
Method
mass effusion(total U)
mass effusion(total U)
Langmuir
mass effusion(total U)
mass effusion(total U)
transpiration
mass effusion(total U)
mass spect.UO2(g)
transpiration
Temp. Range(K)
1758-2378(1600-2809)
1930-2160
1723-2573
2200-2800
2021-2963
2090-2900
1873-2573
1890-2420
2080-2705
AH(kcai/mole)
142.5+1.4
150.0+4.0
140.5+1.4
151.8+3.3
108.1
140.9+1.9
147.5+0.5
141.2+1.1
143.1+3.0
AS(cal/mol/deg)
39.2+0.7
43.6+1.9
36.8+0.2
43.7+1.4
26.3
37.9+0.8
42.5+0.5
39.4+0.5
39.4+1.2
Vapor Pressureat 2150K (x 106 atm)
1.2,
1.9/
0.57?
1.32
*5.75
0.914
1.97
1.8,
1 J6
+A recalculation of the tabulated data yields the present values. av. = 1.36+ 0.1g
*Not included in the average
**Systematic temperature corrections obtained from C. A. Alexander were applied to the originally reported valuesand the lowest temperature point has been disregarded.
CO

19
References
[1] R. J. Ackermann, P. W. Gilles, and R. J. Thorn, J. Chem.
Phys., 2J5 (1956) 1089; see also R. J. Ackermann, Argonne
National Laboratory Report, ANL-5482, September, 1955.
[2] V. E. Ivanov, A. A. Krugl, V. S. Pavlov, G. P. Kovtin,
and V. M. Amonenko, in Thermo'dynamics of Nuclear1 Materials,
I.A.E.A., Vienna (1962) 735.
[3] N. M. Voronov, A. S. Danilin, and I. T. Kovalev, ibid.,
p. 789.
t'4] R. W. Ohse, J. Chem. Phys., £4 (1966) 1375.
[5] B. Gross, Institut fiir Kerntechnik der Technischen Universitat
Berlin Report, TUBIK-4, October, 1966.
[6] C. A. Alexander, J. S. Ogden, and G. C. Cunningham, Battelle
Memorial Institute Report, BMI-1789, January, 1967.
[7] Yu. A. Gorban, L. V. Pavlinov and V. N. Bykov, At. Energ. ^2
(1967) 465.
[8] A. Pattoret, J. Drowart and S. Smoes, in Thermodynamics of
Nuclear Materials, 1967, I.A.E.A., Vienna (1968) 613.
[9] M. Tetenbaum and P. D. Hunt, J. Nucl. Mater., 3£ (1970) 86.
[10] R. J. Ackermann, M. S. Chandrasckharaiah and E. G. Rauh,
Argonne National Laboratory Report, ANL-7048, July, 1965.
[11] M. Bober, W. Breitung, H. U. Karow and K. Schretzmann,
J. Nucl. Mater., 6£ (1976) 20.

20
[12] R. W. Ohse, J. F. Babelot, G. D. Brumme, and P. R. Kinsman,
Ber. der Buns. Gesell., 80 (1976) 780.
[13] N. Asami, M. Nishikawa, and M. Tagucki, in Thermodynamics of
Huclear Materials, 1974, I.A.E.A., Vienna (1975), Vol. 1,
p. 287.
[14] K. D. Carlson, P. W. Gilles and R. J. Thorn, J. Chem. Phys.,
3_8 (1963) 2725.
[15] D. A. Schulz and A. W. Searcy, J. Phys. Chem., 62 (1963) 103.
[16] C. T. Ewing and K. H. Stern, J. Phys. Chem., 7J3 (1974) 1998.
[17] E. G. Rauh, R. C. Sadler, and R. J. Thorn, Argonne National
Laboratory Report, ANL-6536, April, 1962.
[18] H. H. Cremer, Z. Instrumentenkd., 1_ (1964) 72.
[19] See for a description of the equipment and procedure,
R. J. Thorn and G. H. Winslow, Am. Soa. Meoh. Eng. Pap.,
63-WA, 244 (1963).
[20] R. J. Ackermann, unpublished work.
[21] B. F. Naylor, J. Am. Chem. Soc, 61_ (1945) 150.
[22] A. S. Dworkin and M. A. Bredig, J. Chem. Eng. Data, j} (1963)
41.
[23] M. A. Bredig in Colloq. Int. sur I'Etude des Trans. Cryst. a
Haute Temp, au-dessus de 2000°K, Odeillo, Septemhar 27-30, 1971,
C.N.R.S., Paris (1972) 183-191.

21
[24] M. H. Rand, R. J. Ackermann, F. Grtfnvold, F. L. Oetting,
and A. Pattoret, Rev. Int. Hautes Temp, et Refractaires,
to appear.
[25] L. Brewer and M. S. Chandrasekharaiah, University of
California, Radiation Laboratory Report, UCRL-8713 (Revised)
(1960)
[26] L. Brewer and G. M. Rosenblatt, in Advances in High Temperature
Chemistry, (L. Eyring, ed.) 2_, 1-83 (1969).
[27] R. P. Steiger and E. David Cater, High Temperature Science,
1 (1975) 288.
[28] R. J, Ackermann and E. G. Rauh, J. Pure and Appl. Chem., to
appear.
[29] R. J. Ackermann, E. G. Rauh and M. S. Chandrasekharaia,
Argonne National Laboratory Report, ANL-7048, July, 1965.
[30] Leo Brewer, J. Opt. Soc. America, §1 (1971) 1666.
[31] E. D. Cater, Argonne National Laboratory Report, ANL-6140,
March, 1960, page 86.
[32] E. A. Fischer, P. R. Kinsman and R. W. Ohse, J. Nucl. Mater.,
59 (1975) 125.

22
FIGURE CAPTIONS
Fig. 1. Mass-spectrometric measurement of temperature dependence
of p(UO2) over UO2(s), log I(UO2)T vs 104/T.
Fig. 2. Residuals from least squares analyses of mass-spectrometric
results of Fig. 1. Upper - from linear least squares,
log I(UO2)T = A + B/T. Lower - from three-parameter
least squares, log I(UO2)T = A + B/T + C en T.
Fig. 3. C (UO2,g), C (UO2#s) and AC vs T.
Fig, 4. Comparison of the partial pressure of UO2(g) over UO2(s)
from this study with total pressures of uranium bearing
species of other investigations. 0 Ackermann et al.,
Ref. 1, Series B-4 and B-8. • Ibid, Series H-l and H-2.
X , Ohse, Ref. 12. • Cater, Ref. 31. © This study.
Tetenbaum and Hunt, Ref. 9. p(UO2), equation
(1), this study.
Fig. 5. Departure from molecular flow at high temperatures.
• - Hg, Carlson, Ref. 14. © - U02, Ackermann et al.,
Ref. 1, Series B-4. A U02, Ohse, Ref. 12.
COro

3
y= !
oq I
(cal
c),
i
en"
3a
RES
II
+.05
, o
-.05
+.05
0
-.05
—o
o°
o
oo
©
o
0
o o
© 0
o
o
1
oo
° o
«
0
oo°© '
o
©
1
o
0
o
*
>
o
o°
1
o
»*©*<
0° °
1
1
» ° © •
• 1 ° •••©0 O
8© °et © ©
o •
o
8 ©o ©
o o o
o ••
1
0
f
©
>
8°
o
o
••v.0
0
°
1
1
©
o
o oo
o
O*©o oo
o
«
© o
° 00
o
« -8© o
0
© 00
1
©o
°© o
oo
o
oo
o ©
o
<
o
0
©
©
1
•
0 <o
0
o
o
1
—9
0
o
—
©o
©
1800 1900 2000 2100 2200T6K
2300 2400 2500

6ap/|Oiii/|DO(slZon)dO
6ap/|0Ui/|D3 (6*2o n) d0

HUD/ d 6o|

t?
2.0
1.5
1.0
O
b
0.0011
O A- Q O W-
1
PRESSURE (torr)0.01 O.I
1
X/c
i
* l
o
AA
r
& °o o
m mm
i I
11
o
o
JA
•
1-6.0 -5.0" -4.0
log ps/atm-3.0
- 2 . 0
1.5
0
-2.0