context-speciï¬c requirements for fgfr1 signaling through frs2
TRANSCRIPT

DEVELO
PMENT
663RESEARCH ARTICLE
INTRODUCTIONMembers of the receptor tyrosine kinase (RTK) superfamily elicitsimilar cellular, transcriptional and biochemical responses whenstimulated in cultured cells (Fambrough et al., 1999; Schlessinger,2000), yet direct specific, non-redundant cellular responses indeveloping organisms. It has been proposed that qualitativedifferences in signal transduction underlie RTK functionalspecialization. Such differences could be in ligand sensitivity,effector pathway use or the strength or duration of signaling. Thismodel has gained support from in vivo domain swap experiments,which have demonstrated biologically significant differences in RTKsignal transduction (Klinghoffer et al., 2001; Madhani, 2001;Hamilton et al., 2003). Analyses of mice harboring signaling domainmutations have also shown that effector pathways are differentiallyrequired within the RTK superfamily, and that many signaling eventsobserved in biochemical studies are dispensable or required only fora subset of the in vivo functions of receptors (Partanen et al., 1998;Tallquist et al., 2000; Maina et al., 2001; Klinghoffer et al., 2002;Tallquist et al., 2003).
The mammalian fibroblast growth factor (Fgf) signaling networkconsists of four high affinity RTKs and at least 22 ligands (Ornitz,2000; Böttcher and Niehrs, 2005). Mouse knockout studies haveidentified numerous roles of Fgfs, and demonstrated that all essentialembryonic roles are mediated by two receptors, Fgfr1 and Fgfr2(Deng et al., 1994; Yamaguchi et al., 1994; Deng et al., 1996; Armanet al., 1998; Yu et al., 2000; Böttcher and Niehrs, 2005). We havefocused our studies on Fgfr1, which is required for postimplantationgrowth, mesodermal migration and patterning during gastrulation,and for somitogenesis. Fgfr1null mouse embryos also exhibitposterior truncations and neural tube closure defects (Deng et al.,1994; Yamaguchi et al., 1994). Studies of hypomorphic andconditional alleles have identified additional roles of this receptor innode regression, neural stem cells, olfactory bulb development, and
patterning of the anteroposterior axis, central nervous system andpharyngeal arches (Partanen et al., 1998; Tropepe et al., 1999; Xu etal., 1999; Hébert et al., 2003; Trokovic et al., 2003a; Trokovic et al.,2003b).
Though many roles of Fgfr1 are known, the mechanisms bywhich this receptor signals in vivo have not yet been elucidated.Active Fgfrs can directly engage relatively few proteins, namely Crk,Grb14, Shb, PLC� and Frs2,3 (hereafter referred to collectively as‘Frs’) (Mohammadi et al., 1991; Wang et al., 1996; Kouhara et al.,1997; Larrson et al., 1999; Reilly et al., 2000; Cross et al., 2002).Biochemical studies have implicated Frs adaptors as the keymediators of Fgfr signal transduction. These proteins interactconstitutively with the intracellular juxtamembrane regions of Fgfr1and Fgfr2, unlike signaling proteins downstream of other RTKs thatare recruited after receptor activation. Fgfr activation leads tophosphorylation of Frs tyrosine residues to which Grb2 and SHP2are subsequently recruited, initiating PI3K and MAPK signaling(Wang et al., 1996; Kouhara et al., 1997; Xu et al., 1998; Ong et al.,2000; Hadari et al., 2001). This adaptor-mediated mechanism maydistinguish the kinetics, amplitude or subcellular localization of Fgfrsignal transduction from other RTK signaling events.
Frs adaptors are conserved among vertebrates, and expressionpatterns suggest that the two mammalian isoforms perform non-redundant functions in vivo (McDougall et al., 2001; Gotoh et al.,2004). Loss-of-function studies have not been reported for Frs3,but Frs2–/– mouse embryos die ~E7.5 with defects in extra-embryonic development (Hadari et al., 2001; Gotoh et al., 2005).Mouse chimera and Xenopus knockdown studies have furtherimplicated Frs2 in mesoderm development and convergentextension, respectively (Akagi et al., 2002; Gotoh et al., 2005).These studies are informative with respect to Frs2 roles, but theyare difficult to interpret in terms of specific growth factor pathwaysbecause of the promiscuity of Frs adaptors. In addition to Fgfrs,Frs can interact with active neurotrophin receptors and arephosphorylated downstream of other RTKs (Rabin et al., 1993;Ong et al., 2000). Furthermore, both adaptors associate stronglywith the cell cycle regulatory protein p13suc1, and may thus play arole in cell cycle regulation or progression (Rabin et al., 1993; Onget al., 1996).
Context-specific requirements for Fgfr1 signaling throughFrs2 and Frs3 during mouse developmentRenée V. Hoch and Philippe Soriano*
Fibroblast growth factor receptor 1 (Fgfr1) plays pleiotropic roles during embryonic development, but the mechanisms by which thisreceptor signals in vivo have not previously been elucidated. Biochemical studies have implicated Fgf receptor-specific substrates(Frs2, Frs3) as the principal mediators of Fgfr1 signal transduction to the MAPK and PI3K pathways. To determine thedevelopmental requirements for Fgfr1-Frs signaling, we generated mice (Fgfr1�Frs/�Frs) in which the Frs2/3-binding site on Fgfr1 isdeleted. Fgfr1�Frs/�Frs embryos die during late embryogenesis, and exhibit defects in neural tube closure and in the development ofthe tail bud and pharyngeal arches. However, the mutant receptor is able to drive Fgfr1 functions during gastrulation andsomitogenesis, and drives normal MAPK responses to Fgf. These findings indicate that Fgfr1 uses distinct signal transductionmechanisms in different developmental contexts, and that some essential functions of this receptor are mediated by Frs-independent signaling.
KEY WORDS: Fgfr1, Frs2, Frs3, Signaling, Gastrulation, Neural tube, Tail bud, Pharyngeal arches
Development 133, 663-673 doi:10.1242/dev.02242
Program in Developmental Biology, Division of Basic Sciences, Fred HutchinsonCancer Research Center, 1100 Fairview Avenue North, Seattle, WA 98109, USA.
*Author for correspondence (e-mail: [email protected])
Accepted 8 December 2005

DEVELO
PMENT
664
To assess the developmental requirements for Frs-mediatedsignaling downstream of Fgfr1, we have generated mice lacking theFrs-binding site on this receptor (Fgfr1�Frs). We found that Frs-mediated signaling is dispensable for Fgfr1 functions duringgastrulation and somitogenesis, but is required for Fgfr1 roles inneurulation, tail bud and pharyngeal arch development. Furthermore,we found that primary embryonic cells do not require this signalinginteraction to elicit strong and sustained MAPK responses to Fgf, andthat MAPK activation in vivo is not grossly affected by the Fgfr1�Frs
mutation. These results indicate that Frs adaptors are not the exclusiveeffectors of Fgfr1 signal transduction in vivo.
MATERIALS AND METHODSTargeting vector constructionTargeting vectors were generated in pPGKneoF2L2DTA, a plasmidcontaining a DTA selection cassette and a neomycin cassette flanked bynested loxP and FRT sites. For knock-in (KI) vectors (Fgfr1wtKI, Fgfr1�Frs),the short arm of homology, an XbaI/HindIII fragment spanning introns 6-7of Fgfr1, was inserted upstream of the 5� loxP site in the same orientation asthe neomycin cassette. Intron 7-partial cDNA cassettes were generatedthrough two rounds of PCR SOEing (splicing by overhang extension, oligosare given in Table S1 in the supplementary material) (Pogulis et al., 2000).In the first SOEing reaction, nucleotides 1276-1356 (amino acids 407-433)were deleted from a mouse Fgfr1 cDNA construct to generate an Fgfr1�Frs
cDNA. In the second round, partial Fgfr1wt, Fgfr1�Frs cDNAs (exons 8-17)were spliced to the 3� end of intron 7, preserving the endogenous intron7/exon 8 junction. All PCR products incorporated into targeting vectors weregenerated using Pfu (Stratagene) and were verified by sequencing. Intron 7-partial cDNA cassettes were digested with Tth111I and ligated to the longarm of homology, a ~4.1 kb Tth111I/PstI genomic fragment. Resultantfragments were cloned between the 3� loxP site and DTA (Fig. 1B).
For the Fgfr1floxex4 vector, the short arm of homology, exon 4 fragment,and part of the 3� long arm of homology were PCR amplified from wild type129Sv genomic DNA (oligos are given in Table S1 in the supplementarymaterial). The exon 4 fragment was cloned between loxP sites, upstream ofthe FRT-flanked neomycin cassette in pPGKneoF2L2DTA. The short armwas inserted upstream of the 5� loxP site; the intron 5 PCR fragment wasligated to a genomic HindIII/EcoRV fragment to complete the long arm,which was inserted between the second loxP site and DTA (Fig. 1C).
Generation of mouse linesAK7 ES cells were electroporated with linearized targeting vectors, G418selected and PCR screened for integration at the Fgfr1 locus (data notshown). Targeting was verified by Southern analysis with the followingprobes: Fgfr1KI–5� external (NcoI/XmaI), internal (SpeI/NciI), 3� external(PstI/HindIII); Fgfr1�ex4–5� external (PCR product from 129Sv genomicDNA, oligos are given in Table S1 in the supplementary material); internal(NotI/NcoI).
Correctly targeted ES cells were injected into wild type C57BL/6Jblastocysts to generate chimeras. The neomycin cassette caused earlyembryonic lethality of KI homozygotes and was excised through breedingto Meox2-Cre or FlpeR mice (Farley et al., 2000; Tallquist and Soriano,2000). Complete excision of neomycin (Fgfr1KI, Fgfr1floxex4) and exon 4(Fgfr1floxex4) was verified by Southern analysis (data not shown). Tailbiopsies were routinely PCR genotyped (oligos are given in Table S1 in thesupplementary material). In KI homozygotes, embryonic RT-PCR confirmedthat targeting did not disrupt alternative splicing of the extracellular domainor splicing of upstream exons into the cDNA (data not shown).
Whole-mount in situ hybridizationWhole-mount in situ hybridization was performed using standardprocedures.
RNA preparation and semi-quantitative RT-PCREmbryonic RNA was prepared using Trizol (GibcoBRL) and reversetranscribed as described previously (Chen and Soriano, 2003). DilutedcDNA pools were amplified using the following primer pairs:FGFR1.52/FGFR1.36, FGFR2ex11.51/FGFR2ex13.31, Timm_cDNA+/
Timm_cDNA- (see Table S1 in the supplementary material). TriplicatePCR reactions were cycled within a linear range of amplification andproducts were detected and quantified using Sybr Gold (Molecular Probes)and NIH ImageQuant software. Fgfr1 and Fgfr2 RT-PCR products werenormalized to Timm (mitochondrial membrane transport protein) levels.
ImmunohistochemistryWhole-mount phospho-MAPK IHC was performed as described previously(Corson et al., 2003), with minor modifications.
For thin section immunohistochemistry, embryos were fixed overnight(4% PFA), embedded in paraffin wax and sectioned. After rehydration,slides were pretreated in (3% H2O2, 10% methanol, 1�PBS). Antigenretrieval was performed by microwaving slides in 10 mM citrate buffer (pH6.0) (phosphohistone H3, Upstate Biotechnology #06-570) or 10 mM EDTA(pH 8.0) (pMAPK, Cell Signaling #9101S). Samples were blocked in 5%serum/PBS and incubated with antibody overnight. Immunocomplexes weredetected using biotinylated secondary antibodies with ABC Elite and DABsubstrate kits (Vectastain).
Nile Blue and TUNEL analysisFreshly isolated embryos were stained in Nile Blue (0.003% in DMEM,0.5% FBS) for 30-60 minutes (37°C) and washed in PBS. For TUNELanalysis, embryos were fixed, embedded and sectioned as forimmunohistochemistry. Rehydrated sections were incubated in (0.3%Triton-X-100, 1x PBS), digested with proteinase K, and incubated in TdTreaction mix for 1 hour, 37°C [30 mM Tris pH 7.5, 140 mM cacodylate, 1mM CoCl2, 10 �M dig-dUTP, 0.3 U/ �L TdT (Roche)]. Reactions werestopped in PBS and TUNEL-positive nuclei were visualized by IHC withanti-Dig-AP Fab (Roche) and NBT/BCIP detection.
Skeletal preparationsSkeletons were prepared and stained with Alcian Blue and Alizarin Redusing a standard protocol.
Mouse embryonic cell (MEC) isolation and cultureE12.5-14.5 embryos were trypsinized (5�, 37°C), gently disaggregated witha Pasteur pipette, and plated on gelatin in DMEM, 15% FBS. To generateimmortalized lines, p1 MECs were infected with an E6LTTNLoxPretrovirus transducing the SV40 Large T antigen (Berghella et al., 1999).Infected cells were selected with 500 �g/ml G418 and resistant colonieswere pooled.
Immunoprecipitation (IPs) and western blottingMECs were seeded at 1.5-1.8�104 cells/cm2, serum starved (24-36 hours,0.5% FBS) and stimulated with heparin and Fgf (aFgf and bFgf gave similarresults; Research Diagnostics). Cells were then washed with cold PBS andlysed in HNTG (20 mM HEPES pH 7.9, 150 mM NaCl, 1% Triton X-100,10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1 �g/ml aprotinin, 1 mM PMSF,10 mM NaPPi, 0.2 mM activated Na2VO3, 50 mM NaF). IPs were performedovernight in HNTG; immunocomplexes were pulled down using protein A-or G-PLUS agarose (Santa Cruz) and washed in HNTG or (500 mM NaCl, 10mM Tris pH 7.5, 2 mM EDTA, 1% NP40). SDS-PAGE and western blottingwere performed according to standard protocols, with phospho-specific and -nonspecific antibody blots blocked in 1% BSA and 3% milk, respectively.
Antibodies: pMAPK (Cell Signaling); Frs2 (Santa Cruz H-91); Fgfr2(mouse monoclonal, Research Diagnostics); RasGAP (70.3; gift of AndriusKazlauskas; Valius et al., 1993); phosphotyrosine (clone 4G10, UpstateBiotechnology); pAkt (Cell Signaling); Crk (BD Transduction Labs); andactin (clone AC-15, Sigma).
RESULTSGeneration of Fgfr1��Frs and Fgfr1wtKI miceWe used a partial cDNA knock-in approach to generate mice lackingthe Frs binding site on Fgfr1 without disrupting alternative splicingof exons encoding the extracellular domain (Fig. 1) (Werner et al.,1992; Yan et al., 1992; Johnson and Williams, 1993). This deletionmutation has previously been shown to abrogate Fgfr1 binding toboth Frs2 and Frs3 without disrupting other receptor functions and
RESEARCH ARTICLE Development 133 (4)

DEVELO
PMENT
665RESEARCH ARTICLEFgfr1-Frs signaling during mouse development
B
exons 1-7 exons 8-176 7 8 9 10 11 12 13 17pA
FRT siteLoxP siteSouthern probe
Fgfr1 locus(exons 6-17)
Fgfr1wtKI
Fgfr1ΔFrs DTA
3 4 5 6 7C Fgfr1 locus (exons 3-7)
Fgfr1floxex4
Fgfr1Δex4
(+ Cre)
3 5 6 7
7 654 3
PTHX
F8 17pA
vRA dNAhN
neoR cDNA
F neoR F
F
F
DTA
Fgfr1Δex4
(null) Fgfr1ΔFrs
(Δ aa407-433)Fgfr1wtKI
(Wild type KI)
Crk
PLCγ
Frs2/3Crk
PLCγ
A D
Int
E
0
0.2
0.4
0.6
0.8
1
1.2
1.4
+/+
+/+
+/+
wtK
I
wtK
I
wtK
I
wtK
I
wtK
I
Frs
Frs
+/+
+/+
+/+
Frs
Frs
Fg
fr1/
Tim
m (
a.u
.)
5.9E5.11E
Fig. 1. Fgfr1 targeting. (A) Alleles introduced by gene targeting. (B) Partial cDNA knock-in approach: Fgfr1 exons 8-17, encoding thetransmembrane and cytoplasmic domains, were replaced with a pseudoexon encoding the corresponding region of Fgfr1wtKI or Fgfr1�Frs. PartialcDNAs were spliced at the 5� end into exon 8 and at the 3� end into exon 17, upstream of the endogenous polyadenylation sequence. (C) Togenerate the null allele, the first exon common to all reported Fgfr1 splice variants (exon 4) was flanked with loxP sites and excised in vivo byMeox2-Cre (Tallquist and Soriano, 2000). RT-PCR from embryonic RNA confirmed the generation of a stop codon soon after the exon3-exon5 splicejunction (data not shown). (D) Southern blots verifying correct targeting of all alleles in ES cell clones. Abbreviations: A, ApaI; H, HindIII; KI, knock-in; Nd, NdeI; Nh, NheI; P, PstI; Rv, EcoRV; T, Tth111I; X, XbaI; ext, external; int, internal. (E) Relative Fgfr1 mRNA levels in homozygous embryos,assessed by semi-quantitative RT-PCR. Each point is the mean of triplicate reactions for a single embryo (±s.d.).

DEVELO
PMENT
666
interactions (Xu et al., 1998). As a control, we used the sameapproach to generate wild-type knock-in mice (Fgfr1wtKI; theuntargeted allele is designated Fgfr1+).
Fgfr1wtKI/wtKI and Fgfr1��ex4/��ex4 phenotypes on ahigh percentage C57BL/6J backgroundFollowing two or more backcross generations to the C57BL/6Jbackground, we recovered nearly the expected ratio of Fgfr1wtKI/wtKI
animals at E16.5, and a number of homozygotes survived toadulthood with normal fertility (Table 1). We therefore selected thisbackground (>75% contribution from C57BL/6J) for our studies. Onthis genetic background, Fgfr1wtKI/wtKI embryos are often smallerthan littermates and exhibit digit 3/4 fusions (data not shown).Importantly, however, Fgfr1�Frs/�Frs phenotypes discussed below areeither absent or notably less penetrant/severe in Fgfr1wtKI/wtKI
embryos (see Fig. S1 in the supplementary material). Phenotypicdifferences between the knock in lines were not due to differencesin Fgfr1 expression: Fgfr1 mRNA levels are similar in untargeted,Fgfr1wtKI/wtKI and Fgfr1�Frs/�Frs embryos on the high percentage C57background (Fig. 1E).
Because the Fgfr1wtKI/wtKI phenotype is background dependent(data not shown), we revisited the null phenotype on the (>75%)C57BL/6J background. We generated mice harboring a null allele(Fgfr1�ex4, Fig. 1A,C) and backcrossed them more than twogenerations to C57BL/6J. Compared with published Fgfr1null
embryos, which were analyzed on different mixed backgrounds,
Fgfr1�ex4/�ex4 embryos on the (>75%) C57BL/6J backgroundsurvive slightly later in embryogenesis (E11.5 vs. E7.5-9.5).However, they are always developmentally arrested prior to E9.5 andexhibit phenotypes similar to published Fgfr1null embryos, includinga developmental delay, mesodermal migration and patterningdefects, craniorachischisis and posterior truncations (Fig. 2; Fig. 3B)(Deng et al., 1994; Yamaguchi et al., 1994). Fgfr1�ex4/�ex4 embryosalso have enlarged hearts with disrupted looping (data not shown).
Fgfr1-Frs signaling is not required duringgastrulation or somitogenesisFgfr1�Frs/+ animals are viable and indistinguishable from littermatecontrols, but Fgfr1�Frs/�Frs mice were never recovered postnatally(Table 1). Timed matings indicated that Fgfr1�Frs/�Frs embryossurvive much later in embryogenesis than do Fgfr1�ex4/�ex4 embryos,with a drop in viability only after E15.5 (Fig. 2A).
Fgfr1 roles in early mesodermal development were rescued in~80% of Fgfr1�Frs/�Frs embryos. During gastrulation, Fgfr1 isrequired for mesodermal migration through the primitive streak. InFgfr1�ex4/�ex4 (and published null) embryos, this migration isimpaired and cells accumulate in the streak. Consequently, there isan expansion of axial mesoderm and reduction of paraxial mesoderm,which remains disorganized and fails to form somites (Fig. 2B,C)(Deng et al., 1994; Yamaguchi et al., 1994; Ciruna et al., 1997; Cirunaand Rossant, 2001). We examined the development of axial andparaxial mesoderm in Fgfr1�Frs/�Frs embryos by Shh and Meox1 insitu hybridizations, respectively. Although Fgfr1�Frs/�Frs embryos aredevelopmentally delayed by ~1 day, most mutants exhibit normalexpression of both mesodermal markers following gastrulation (Fig.2D-G). This indicates that mesodermal migration is rescued in theseembryos. Furthermore, whereas Fgfr1�ex4/�ex4 embryos never formsomites, Fgfr1�Frs/�Frs embryos undergo normal somitogenesis, asevidenced by the segmented pattern of Meox1 staining.
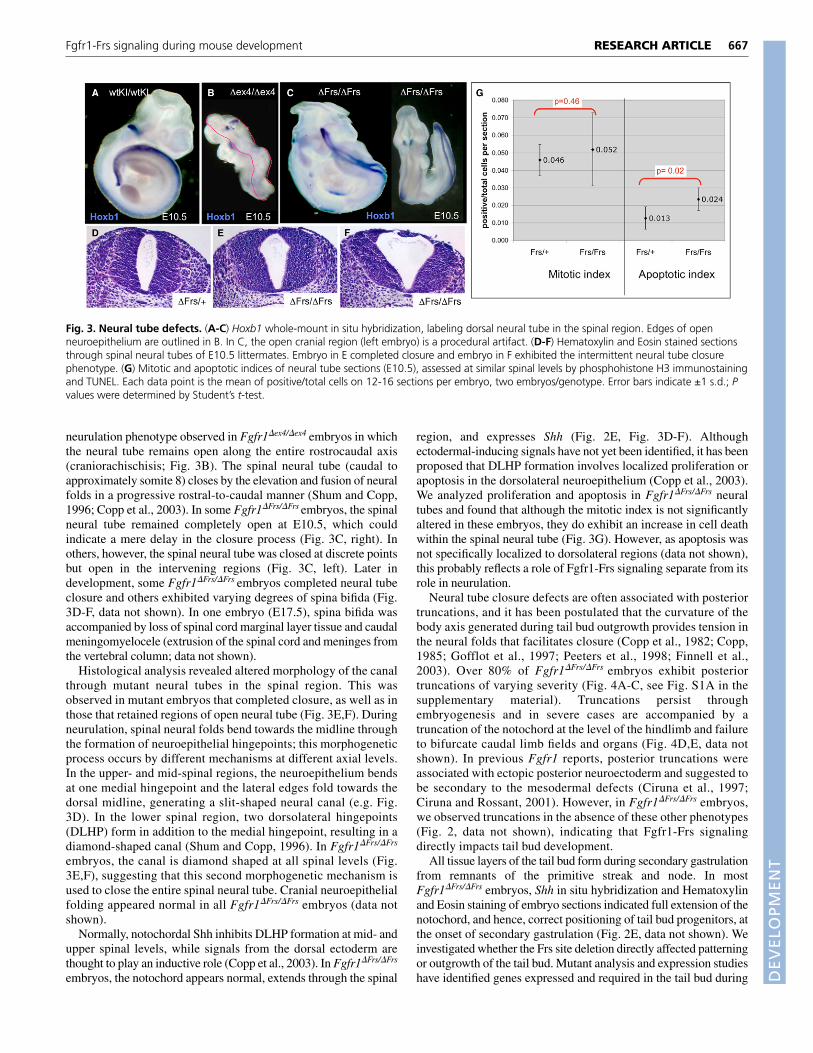
Fgfr1-Frs signaling is essential during neurulationand tail bud developmentThirty to 50% of Fgfr1�Frs/�Frs embryos isolated E10.5-11.5exhibited a defect in spinal neural tube closure (Fig. 3A,C). This wasnever observed in Fgfr1wtKI/wtKI embryos, and differs from the
RESEARCH ARTICLE Development 133 (4)
Table 1. Genotypes of offspring from heterozygote knock-in(KI) intercrosses at late embryonic and postnatal stagesAge Line n +/+ (%) +/KI (%) KI/KI (%)
P4-P7 wtKI #1 72 24 (33) 36 (50) 12 (17)wtKI #2 31 9 (29) 20 (65) 2 (6)
�Frs 60 18 (30) 42 (70) 0
E16.5 wtKI #2 24 7 (29) 13 (54) 4 (17)wtKI #3 17 4 (24) 10 (59) 3 (18)
�Frs 22 4 (18) 17 (77) 1 (5)
wtKI #1-3 are KI mouse lines derived from independently targeted ES cell clones.
Fig. 2. Rescue of early Fgfr1 functions by Fgfr1��Frs.(A) Viability (percent of expected) of homozygotesrecovered from Fgfr1�Frs/+ and Fgfr1�ex4/+ intercrosses.(B-G) Sonic hedgehog (Shh, E9.5) and Meox1 (E10.5) insitu hybridization. Arrows indicate width of the Shhexpression domain (more than two cell diameters inFgfr1�ex4/�ex4 compared with one cell diameter in controlembryos). (C) The paraxial mesoderm population isreduced and disorganized in Fgfr1�ex4/�ex4 embryos, soMeox1 staining is faint and diffuse. (Color developmenttime required to visualize any positive signal was notablylonger in this embryo than in F,G.)
DEVELO
PMENT
neurulation phenotype observed in Fgfr1�ex4/�ex4 embryos in whichthe neural tube remains open along the entire rostrocaudal axis(craniorachischisis; Fig. 3B). The spinal neural tube (caudal toapproximately somite 8) closes by the elevation and fusion of neuralfolds in a progressive rostral-to-caudal manner (Shum and Copp,1996; Copp et al., 2003). In some Fgfr1�Frs/�Frs embryos, the spinalneural tube remained completely open at E10.5, which couldindicate a mere delay in the closure process (Fig. 3C, right). Inothers, however, the spinal neural tube was closed at discrete pointsbut open in the intervening regions (Fig. 3C, left). Later indevelopment, some Fgfr1�Frs/�Frs embryos completed neural tubeclosure and others exhibited varying degrees of spina bifida (Fig.3D-F, data not shown). In one embryo (E17.5), spina bifida wasaccompanied by loss of spinal cord marginal layer tissue and caudalmeningomyelocele (extrusion of the spinal cord and meninges fromthe vertebral column; data not shown).
Histological analysis revealed altered morphology of the canalthrough mutant neural tubes in the spinal region. This wasobserved in mutant embryos that completed closure, as well as inthose that retained regions of open neural tube (Fig. 3E,F). Duringneurulation, spinal neural folds bend towards the midline throughthe formation of neuroepithelial hingepoints; this morphogeneticprocess occurs by different mechanisms at different axial levels.In the upper- and mid-spinal regions, the neuroepithelium bendsat one medial hingepoint and the lateral edges fold towards thedorsal midline, generating a slit-shaped neural canal (e.g. Fig.3D). In the lower spinal region, two dorsolateral hingepoints(DLHP) form in addition to the medial hingepoint, resulting in adiamond-shaped canal (Shum and Copp, 1996). In Fgfr1�Frs/�Frs
embryos, the canal is diamond shaped at all spinal levels (Fig.3E,F), suggesting that this second morphogenetic mechanism isused to close the entire spinal neural tube. Cranial neuroepithelialfolding appeared normal in all Fgfr1�Frs/�Frs embryos (data notshown).
Normally, notochordal Shh inhibits DLHP formation at mid- andupper spinal levels, while signals from the dorsal ectoderm arethought to play an inductive role (Copp et al., 2003). In Fgfr1�Frs/�Frs
embryos, the notochord appears normal, extends through the spinal
region, and expresses Shh (Fig. 2E, Fig. 3D-F). Althoughectodermal-inducing signals have not yet been identified, it has beenproposed that DLHP formation involves localized proliferation orapoptosis in the dorsolateral neuroepithelium (Copp et al., 2003).We analyzed proliferation and apoptosis in Fgfr1�Frs/�Frs neuraltubes and found that although the mitotic index is not significantlyaltered in these embryos, they do exhibit an increase in cell deathwithin the spinal neural tube (Fig. 3G). However, as apoptosis wasnot specifically localized to dorsolateral regions (data not shown),this probably reflects a role of Fgfr1-Frs signaling separate from itsrole in neurulation.
Neural tube closure defects are often associated with posteriortruncations, and it has been postulated that the curvature of thebody axis generated during tail bud outgrowth provides tension inthe neural folds that facilitates closure (Copp et al., 1982; Copp,1985; Gofflot et al., 1997; Peeters et al., 1998; Finnell et al.,2003). Over 80% of Fgfr1�Frs/�Frs embryos exhibit posteriortruncations of varying severity (Fig. 4A-C, see Fig. S1A in thesupplementary material). Truncations persist throughembryogenesis and in severe cases are accompanied by atruncation of the notochord at the level of the hindlimb and failureto bifurcate caudal limb fields and organs (Fig. 4D,E, data notshown). In previous Fgfr1 reports, posterior truncations wereassociated with ectopic posterior neuroectoderm and suggested tobe secondary to the mesodermal defects (Ciruna et al., 1997;Ciruna and Rossant, 2001). However, in Fgfr1�Frs/�Frs embryos,we observed truncations in the absence of these other phenotypes(Fig. 2, data not shown), indicating that Fgfr1-Frs signalingdirectly impacts tail bud development.
All tissue layers of the tail bud form during secondary gastrulationfrom remnants of the primitive streak and node. In mostFgfr1�Frs/�Frs embryos, Shh in situ hybridization and Hematoxylinand Eosin staining of embryo sections indicated full extension of thenotochord, and hence, correct positioning of tail bud progenitors, atthe onset of secondary gastrulation (Fig. 2E, data not shown). Weinvestigated whether the Frs site deletion directly affected patterningor outgrowth of the tail bud. Mutant analysis and expression studieshave identified genes expressed and required in the tail bud during
667RESEARCH ARTICLEFgfr1-Frs signaling during mouse development
Fig. 3. Neural tube defects. (A-C) Hoxb1 whole-mount in situ hybridization, labeling dorsal neural tube in the spinal region. Edges of openneuroepithelium are outlined in B. In C, the open cranial region (left embryo) is a procedural artifact. (D-F) Hematoxylin and Eosin stained sectionsthrough spinal neural tubes of E10.5 littermates. Embryo in E completed closure and embryo in F exhibited the intermittent neural tube closurephenotype. (G) Mitotic and apoptotic indices of neural tube sections (E10.5), assessed at similar spinal levels by phosphohistone H3 immunostainingand TUNEL. Each data point is the mean of positive/total cells on 12-16 sections per embryo, two embryos/genotype. Error bars indicate ±1 s.d.; Pvalues were determined by Student’s t-test.

DEVELO
PMENT
668
secondary gastrulation (Roelink and Nusse, 1991; Takada et al.,1994; Greco et al., 1996; Gofflot et al., 1997; Copp et al., 2003).Except in the most severely truncated embryos, tail bud geneexpression (T, Wnt3a, Fgf8, Hoxb1) and proliferation appearednormal in Fgfr1�Frs/�Frs tail buds (Fig. 4F-I, data not shown).However, TUNEL analysis revealed an increase in cell deaththroughout caudal tissues of Fgfr1�Frs/�Frs embryos (Fig. 4I). Celldeath may hinder cell migrations or reduce the number of tail budprogenitors during secondary gastrulation.
Fgfr1-Frs signaling in pharyngeal arch (PA)developmentThe second PA is hypoplastic in Fgfr1�Frs/�Frs embryos byE10.5, and histological analysis revealed a dramatic reductionin the number of mesenchymal cells populating this arch. Thispopulation normally includes both mesoderm and neural crestcells. Unlike cells within wild-type PAs, mesenchymal cellswithin mutant PA2 are neither proliferating nor closely apposedto the surrounding epithelia (Fig. 5A-C). We also observed a lossof Fgf8 expression in PA2, which has been shown to promotesurvival of PA mesenchyme (Fig. 5D,E) (Frank et al., 2002;Macatee et al., 2003). Although TUNEL analysis on sections didnot reveal ectopic apoptosis of cells within Fgfr1�Frs/�Frs arches(data not shown), the loss of Fgf8 signals may contribute to thelack of PA2 mesenchymal proliferation or affect migration ofneural crest cells into the arches of mutant embryos. In aprevious study of Fgfr1 hypomorphs, PA2 was found to behypoplastic because of a neural crest migration defect (Trokovicet al., 2003a). Likewise, we observed impaired neural crest cellmigration into Fgfr1�Frs/�Frs PAs by Sox10 in situ hybridization
RESEARCH ARTICLE Development 133 (4)
Fig. 4. Posterior truncations and tail bud development.(A-C) Bright-field photographs of E13.5 littermates (caudal region).(D,E) Hematoxylin and Eosin stained sections through kidneys of wild-type and severely truncated Fgfr1�Frs/�Frs embryos; bar indicates relativemagnification. (F) T (Brachyury) and (G,H) Wnt3a whole-mount in situhybridization. (I) Quantitation of proliferation (pH3, P=0.33) andapoptosis (TUNEL, P=0.09) in E10.5 tail bud sections. Each data pointrepresents the mean of three to eight sections through each of at leastthree embryos. Error bars indicate ±1 s.d.; P values were determined byStudent’s t-test.
Fig. 5. Pharyngeal arch (PA) development. (A-C) Phosphohistone H3immunostained PA sections (E10.5). PA1 is labeled, arrowheads indicatePA2. (D,E) Fgf8 whole-mount in situ hybridization (PA region); arrowsindicate PA2 Fgf8 expression domain lost in mutant embryos.(F-H) Migrating NCCs in the pharyngeal region of stage-matchedembryos, visualized by Sox10 in situ hybridization. Arrows indicatelimits of NCC migration into PA1, 2. (I-L) Nile Blue staining of cell deathin the pharyngeal region (E10.5). I/J and K/L are stage-matchedcontrol/mutant embryo pairs.

DEVELO
PMENT
(Fig. 5F-H) (Kuhlbrodt et al., 1998). Nile Blue staining revealedectopic cell death along the path of neural crest migration (priorto PA entry; Fig. 5I-L).
At later stages, we noted some recovery of PA2 size and did notobserve prominent craniofacial defects that would be expected ifPA neural crest development failed entirely. We therefore analyzedthe formation of PA crest derivatives to determine whether neuralcrest migration recovered later in development. Analysis ofcraniofacial skeletal elements at E15.5 indicated that PA2 NCCderivatives are selectively affected in Fgfr1�Frs/�Frs embryos. We didnot observe major hypoplasia or malformation of the jaws (PA1-derived; data not shown) or PA1 derivatives in the middle ear (Fig.6A-D). By contrast, cartilages derived from PA2, including thestapes and styloid process of the middle ear and the lesser horns ofthe hyoid, were missing or hypoplastic in mutant embryos (Fig. 6).
Fgf signaling responses in Fgfr1��Frs/��Frs primarycellsPhenotypic data demonstrated that Fgfr1�Frs is expressed andfunctional in vivo: we observed neither recapitulation of the nullphenotype, as would be expected if the mutant receptor were non-functional, nor gain-of-function phenotypes indicative of ligand-independent activation. However, phenotypic data also indicated thatFgfr1 transduces Frs-independent signals in some developmentalcontexts. To investigate the nature of these signals, we derivedmouse embryonic cells (MECs) from wild-type and mutantembryos, and used them in biochemical studies.
We first used immortalized MECs to verify that the mutantreceptor signaled as expected based on previous reports. In thesecells, we observed rapid and robust phosphorylation of Frs2 in wild
type, but not Fgfr1�Frs/�Frs, cells stimulated with Fgf (see Fig. S2A,part i in the supplementary material). We also observed an Fgf-stimulated decrease in Frs2 protein level in wild-type but not mutantcells, concomitant with Frs2 phosphorylation (see Fig. 2A, part ii inthe supplementary material). In MAPK response assays, mutantimmortalized MECs exhibited reduced sensitivity to Fgf and droveweaker signaling responses as assessed by phosphorylation ofMAPK (see Fig. S2B in the supplementary material). These datarecapitulate findings of previous studies performed in immortalizedFrs2–/– MECs (Hadari et al., 2001).
Cellular transformation may affect signal transduction and thusconfound results of biochemical studies. Therefore, to approximatethe in vivo situation, we re-examined Fgf responses in low passage(p2) non-immortalized MECs. Surprisingly, Fgfr1�Frs/�Frs and wild-type p2 MECs responded with indistinguishable MAPK activationprofiles, both in terms of response duration and Fgf dose sensitivity(Fig. 7A). We did not observe a notable PI3K pathway response,assayed by Akt phosphorylation, in cells of either genotype (Fig.7A). In addition, neither Crk nor PLC� were tyrosinephosphorylated (i.e. activated) in p2 MECs following Fgfstimulation (Fig. 7B, data not shown). Thus, these pathways areprobably not mediating the observed MAPK response inFgfr1�Frs/�Frs cells. Furthermore, compared with wild-type cells, theFrs2 phosphorylation response was severely diminished in mutantcells despite comparable levels of total Frs2 (Fig. 7C). We expectthat residual Frs activation in Fgfr1�Frs/�Frs cells occurs indirectly,as it has previously been shown by yeast two hybrid that the aminoacid 407-433 deletion completely abolishes the interaction of theFgfr1 cytoplasmic domain with Frs2 (Xu et al., 1998).
We next investigated whether signaling through Fgfr2 couldcompensate for the lack of Fgfr1-Frs signaling by activating Frs2 (lowlevel) and MAPK in Fgfr1�Frs/�Frs MECs. Fgfr2 expression levels arenot altered in Fgfr1�Frs/�Frs (p2) MECs or embryos, as might beexpected if this receptor was upregulated to compensate for reducedFgfr1 signaling (Fig. 7D,E). Furthermore, we analyzed the activationstate (tyrosine phosphorylation) of Fgfr2 in p2 MECs and found thatalthough both wild-type and mutant MECs had basal levels of tyrosinephosphorylated Fgfr2, the amount of active Fgfr2 was not increasedin cells of either genotype in response to Fgf. Surprisingly, the basal(–Fgf) level of activated Fgfr2 was elevated in Fgfr1�Frs/�Frs MECs,and these cells responded to Fgf with a decrease in Fgfr2phosphorylation (Fig. 7D). These data are not consistent with a roleof Fgfr2 in compensatory activation of Frs and MAPK, and insteadsuggest that Fgfr1-Frs signaling transregulates Fgfr2 activity.
The MAPK signaling responses in p2 MECs did not directlytranslate into Fgf responsiveness in a proliferation assay: only twoout of four mutant MEC cultures proliferated in response to Fgf,whereas all four responded with comparable MAPK and PI3Kresponses (Fig. 7A,F, data not shown). The growth curves did,however, reflect the relative phenotypic severity of embryos fromwhich mutant cells were derived (data not shown). Variation inmutant MEC proliferative responses is probably due to differencesin cell physiology or developmental staging between severely andmildly affected embryos. Nonetheless, the ability of two mutantlines to proliferate in response to Fgf indicates that Fgfr1-Frssignaling is not essential in directing this cellular response.
Impact of the Fgfr1��Frs mutation on MAPKsignaling in vivoPrevious reports have implicated the MAPK pathway as an effectorof Fgf signaling in vivo (Corson et al., 2003). Our primary MEC datasupport this model and suggest that the Fgfr1-Frs signaling is not
669RESEARCH ARTICLEFgfr1-Frs signaling during mouse development
Fig. 6. PA2 neural crest derivatives. Skeleton preparations of E15.5middle ears (A,B,D) and hyoid cartilages (E,F). PA1, 2 derivatives aretraced in C,G,H. PA1 neural crest derivatives, traced in blue: i, incus,m, malleus, ty, tympanic ring, mc, Meckel’s cartilage. PA2 derivatives,traced in red, are hypoplastic or missing in mutants: (C) s, stapes, sp,styloid process; (G,H) lesser horns of hyoid. Ectopic cartilages wereobserved in mutant ears (arrows in B,D; yellow in C).

DEVELO
PMENT
670
required for Fgf-induced MAPK activation. To determine thephysiological relevance of the MEC results, we examined MAPKactivation in Fgfr1-dependent developmental contexts by whole-mount immunohistochemistry. First, we analyzed stage-matchedembryos at E8.5-9.5, when the caudal region of the embryo is stillundergoing gastrulation and more rostral axial levels are undergoingsomitogenesis and neurulation. We found that phospho-MAPKstaining in gastrulating mesoderm and somites was not altered inmutant embryos (Fig. 8A-F). In addition, we observed a thin line ofphospho-MAPK staining along the medial edges of the neural folds,where neuroepithelial fusion takes place. This domain of MAPKactivation was present in both mutant and control embryos (arrows,Fig. 8C,F), despite the disruption of neural tube closure inFgfr1�Frs/�Frs embryos. Just after neurulation, when we observed anincrease in cell death in mutant neural tubes, MAPK is not activatedin wild-type or mutant neural tubes (data not shown). Thus, the cellsurvival role of Fgfr1-Frs signaling in the neural tube is probablyfacilitated by a different downstream pathway.
Later, during early PA2 development, phospho-MAPKimmunostained dispersed cells throughout the pharyngeal arch regionin both mutant and control embryos (Fig. 8G,J). As developmentprogressed, phospho-MAPK became concentrated in controlembryos at the rostral edge of PA1 and the caudal edge of PA2 (Fig.
8H). In mutant embryos at this stage, although PA1 immunostainingappeared normal, intensification of phospho-MAPK staining was notobserved in caudal PA2 (Fig. 8K). This domain corresponds to theFgf8 expression domain that is lost in mutants; however, thedifference in MAPK activation observed in whole mount may besecondary to the failure of NCCs to populate this arch. PremigratoryNCCs and NCCs within PA3 stained strongly with thephosphoMAPK antibody (Fig. 8I,L, data not shown).
Together, these results demonstrate that MAPK activity is notglobally disrupted in Fgfr1-dependent contexts in Fgfr1�Frs/�Frs
embryos. Thus, either Fgfr1 does not contribute significantly tocellular phosphoMAPK levels in these contexts, or Fgfr1 signalingto MAPK is not affected by the Frs site deletion.
DISCUSSIONDevelopmental roles of Fgfr1-Frs signalingOur results demonstrate that Fgfr1-Frs signaling is essentialduring embryogenesis but is required only in a subset of Fgfr1-dependent contexts. Abrogation of the Fgfr1-Frs interaction doesnot disrupt Fgfr1 functions during gastrulation or somitogenesis,but does result in late embryonic lethality and defects in thedevelopment of the tail bud, neural tube, and pharyngeal arches.The specific array of phenotypes observed in Fgfr1�Frs/�Frs
RESEARCH ARTICLE Development 133 (4)
Fig. 7. p2 MEC responses to Fgf. (A) pMAPKand pAkt responses of Fgfr1�Frs/�Frs and controlMECs stimulated with Fgf, assayed over a dosetitration (Ai, 5 minute stimulations) and a 1hour timecourse (Aii). RasGAP, loading control.(B,C) Crk and Frs2 activation responses to Fgf(50 ng/ml aFgf, 5 �g/ml heparin, 5�), assayedby IP-phosphotyrosine (4G10) western blot.Blots were stripped and reprobed with anti-Crkor anti-Frs2 (lower panels). *No antibody IPcontrol. (D) Fgfr2 responses to Fgf (50 ng/mlaFgf, 5 �g/ml heparin, 5�), assayed by 4G10 IP-Fgfr2 western blot (upper panel). Total celllysate Fgfr2 (middle panel) and actin (loadingcontrol, lower panel) western blots showrelative protein levels. Matching arrowheadsindicate bands of the same size recognized bydifferent antibodies within B-D. (E) RelativeFgfr2 mRNA levels in homozygous embryos,determined by semi-quantitative RT-PCR.(F) Growth curves of p2 MECs derived fromfour mutant and two wild-type embryoscultured in DMEM, 4% FBS, 50 ng/ml aFgf.

DEVELO
PMENT
embryos is distinct from that observed in Fgfr1wtKI homo- orhemizygotes, previously described hypomorphs with reducedFgfr1 levels, or embryos harboring an Fgfr1PLC� binding sitemutation (Table 2, see Fig. S1 in the supplementary material)(Partanen et al., 1998; Xu et al., 1999; Trokovic et al., 2003a).This suggests that Fgfr1�Frs/�Frs phenotypes are not generalconsequences of reduced Fgfr1 activity, but are indeed due todisrupted signaling through the Frs interaction site. Interestingly,activating mutations in Fgfr1 that cause human craniosynostosissyndromes also have context-specific effects restricted mainly tocraniofacial and limb development (Passos-Bueno et al., 1999).Thus, developmental contexts differ in their requirements forspecific signaling pathways downstream of Fgfr1, as well as forregulation of Fgfr1 activity.
Because Fgfr1�Frs/�Frs embryos exhibited rescue of Fgfr1null
mesodermal phenotypes and early lethality, analysis of thesesignaling mutants enabled us to clarify later roles of Fgfr1 in neuraltube and tail bud development. We thus identified a novel role ofFgfr1 in spinal neurulation. In contrast to craniorachischisis, whichin Fgfr1�ex4/�ex4 embryos is probably secondary to mesodermaldefects, Fgfr1�Frs/�Frs neural tube closure defects affect only thespinal region and are not accompanied by notochord or paraxialmesoderm defects. Our histological data demonstrate that Fgfr1-Frssignaling impacts the morphogenetic folding of the spinalneuroepithelium. Frs2 was previously implicated in convergentextension movements (Akagi et al., 2002), disruption of which canalter the size, shape and signaling properties of the notochord andneural plate, and thus result in neural tube closure defects. However,whereas previously described convergent extension mutants lackmedial hinge points and exhibit notochord defects, Fgfr1�Frs/�Frs
embryos form ectopic DHLPs and do not exhibit defects inmesoderm or medial bending, (Copp et al., 2003). Our results aretherefore more consistent with a role of Fgfr1-Frs signaling inmodulating neuroepithelial responsiveness to DLHP-inducing or-repressing signals.
The neural tube closure defect in Fgfr1�Frs/�Frs embryos may beexacerbated by the accompanying defect in tail bud development.Previously, posterior truncations in Fgfr1null animals were postulatedto be secondary to mesoderm defects. However, the high penetranceof posterior truncations, together with the low incidence ofmesodermal defects in Fgfr1�Frs/�Frs embryos suggest that Fgfr1instead plays a direct, Frs-dependent role in tail bud development.The Frs site deletion did not affect patterning or proliferation in thetail bud during secondary gastrulation, but resulted in ectopicapoptosis throughout the caudal region of mutant embryos. Celldeath could contribute to posterior truncations by depleting tail budprogenitors or hindering cell migration during secondarygastrulation.
In Fgfr1�Frs/�Frs embryos, Sox10-positive neural crest cells arehindered in their migration into the first and second pharyngealarches, though at later stages, craniofacial defects in these embryosare restricted predominantly to PA2 NCC derivatives. This impliesthat the Sox10-positive population migrating towards PA1 recoversor is not essential for the development of some PA1 NCCderivatives. Specific disruption of PA2 and/or PA2 NCCdevelopment in Fgfr1�Frs/�Frs embryos contrasts with general Fgfr1hypomorph phenotypes that affect NCC derivatives of both PA1 andPA2 (Trokovic et al., 2003a). Fgfr1-Frs signaling may be primarilyrequired for PA2 patterning, or may participate in reciprocalsignaling between the epithelium and NCCs that facilitates PA2entry or survival of NCCs. In Fgfr1�Frs/�Frs embryos, an epithelialFgf8 expression domain is lost in PA2, and the NCC migration
671RESEARCH ARTICLEFgfr1-Frs signaling during mouse development
Fig. 8. Phospho-MAPK immunohistochemistry. Whole-mountphospho-MAPK immunohistochemistry of stage-matched embryos:(A-C) E8.5, (D-F) E9.5. High magnification views of (B,E) caudalgastrulating regions and (C,F) more rostral areas undergoingsomitogenesis and neurulation. Arrowheads and arrows indicatephospho-MAPK-positive somites and neural folds, respectively.(G-L) G/J, H/K and I/L are stage-matched embryo pairs (E10.5).(G,H,J,K) Pharyngeal arch 1, 2 region; (I) dorsal and (L) lateral views ofpremigratory neural crest cells (arrows). Lateral view is shown in Lbecause this embryo has an open neural tube.
Table 2. Comparison of Fgfr1 mutant phenotypesFgfr1-dependent context Fgfr1red Fgfr1PLC� Fgfr1�Frs Fgfr1wtKI
GastrulationSomitogenesisNode regression XTail bud development X X (x)Neural tube closure X XAP patterning X XPA NCC development X X (x)Digit development X X X
Fgfr1red, hypomorphic alleles with reduced Fgfr1 expression; X, disrupted inhomozygous embryos; (x) disrupted in Fgfr1wtKI/wtKI embryos but at lowerpenetrance and/or severity than in Fgfr1�Frs/�Frs embryos. See text for references.

DEVELO
PMENT
672
defect is accompanied by an increase in cell death along themigration pathway. Preliminary results of conditional mutagenesisexperiments indicate that ectopic cell death in the pharyngeal regionreflects a NCC-nonautonomous effect of the Frs site deletion (R.V.H.and P.S., unpublished).
Potential mechanisms of Fgfr1 signaling duringdevelopmentIt has long been presumed that Fgfrs signal primarily through theMAPK pathway, and previous biochemical studies implicated Frsadaptors as important mediators of MAPK activation downstreamof Fgfr1. However, we have demonstrated that the Fgfr1-Frsinteraction is dispensable for early developmental roles of Fgfr1 andfor activation of the MAPK pathway, both in primary embryoniccells and in vivo. Wild-type and Fgfr1�Frs/�Frs primary cell responsesto Fgf stimulation were indistinguishable in terms of both dosesensitivity and signaling kinetics, and MAPK phosphorylationappeared normal in several Fgfr1-dependent contexts inFgfr1�Frs/�Frs embryos. Furthermore, proliferative responses of non-immortalized Fgfr1�Frs/�Frs cells suggest that this pathway is notessential in driving Fgf-induced proliferation, although cells fromembryos with more severe phenotypes were growth impaired. Thediscrepancy between our biochemical data and those publishedpreviously probably reflects cell type-specific signalingmechanisms, effects of Frs2 disruption on receptors other thanFgfr1, or effects of cellular transformation on the ability of otherpathways to signal downstream of Fgfr1.
Our work, as well as previous studies of Fgfr1-PLC� signaling,demonstrates that individual signaling events downstream of Fgfr1are required in a context-specific manner during development. Incontexts where Fgfr1 functions are not compromised by the Frs sitedeletion, other pathways may drive cellular responses eitherindependently of or additively with Frs adaptors. Aside from Frs2and Frs3, only PLC�, Crk, Grb14 and Sef have been shown tointeract directly with activated Fgfr1, although Src, STAT and Shcare also activated downstream of Fgfr1 in some cell types(Mohammadi et al., 1991; Klint and Claesson-Welsh, 1999; Larrsonet al., 1999; Reilly et al., 2000; Tsang et al., 2002; Kovalenko et al.,2003). Sensitivity to the Frs site deletion could reflect the availabilityof alternate signaling pathways or the level of overall Fgfr1 signalrequired in a given context, rather than selective use of the Frspathway. Thus, Fgfr1-Frs signaling may be used more broadlyduring development than is revealed by Fgfr1�Frs/�Frs phenotypes.
Fgfr1 and Fgfr2 can form heterodimers in vitro (Bellot et al.,1991). If such heterodimers were to form in vivo, Fgfr1�Frs signalscould get shunted to the Frs pathway downstream of the Fgfr2subunit in Fgfr1�Frs/�Frs embryos. This predicts that heterodimerswould rescue Fgfr1�Frs function in contexts co-expressing Fgfr1and Fgfr2, and that Fgfr2 would be activated by Fgf stimulationin primary cells. Our data do not support this model: Fgfr1�Frs/�Frs
embryos exhibit defects in the tail bud and pharyngeal arches,where Fgfr1 and Fgfr2 are co-expressed, but not duringgastrulation or somitogenesis when the receptors have distinctexpression patterns (Orr-Urtreger et al., 1991; Yamaguchi et al.,1992; Walshe and Mason, 2000). Furthermore, we did not observecompensatory upregulation of Fgfr2 mRNA in vivo, or ligand-dependent activation of Fgfr2 in primary cells. Instead, basallevels of active Fgfr2 are notably elevated in Fgfr1�Frs/�Frs cells,suggesting that Fgfr1-Frs signaling transregulates Fgfr2. This maycoordinate Fgfr responses and/or serve a homeostatic role in vivo.Deregulation of Fgfr2 activity may contribute in a gain-of-
function manner to Fgfr1�Frs/�Frs phenotypes. Resolution of thisissue will require further biochemical analysis and crossesbetween Fgfr1�Frs and Fgfr2 mutant mouse lines.
We thank Janet Rossant for providing Fgfr1 genomic clones; Mitch Goldfarbfor advice on the Frs site deletion; Philip Corrin, Jason Frazier and Marc Grenleyfor excellent technical assistance; and Raj Kapur, Henk Roelink and ourlaboratory colleagues for critical reading of the manuscript. This work wassupported by an NSF predoctoral fellowship and the CMB Training Grant (NIH#GM07270) to R.V.H., and by grants HD 24875 and HD 25326 to P.S.
Supplementary materialSupplementary material for this article is available athttp://dev.biologists.org/cgi/content/full/133/4/663/DC1
ReferencesAkagi, K., Park, E. K., Mood, K. and Daar, I. O. (2002). Docking protein SNT1 is
a critical mediator of fibroblast growth factor signaling during Xenopusembryonic development. Dev. Dyn. 223, 216-228.
Arman, E., Haffner-Krausz, R., Chen, Y., Heath, J. K. and Lonai, P. (1998).Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a rolefor FGF signaling in pregastrulation mammalian development. Proc. Natl. Acad.Sci. USA 95, 5082-5087.
Bellot, F., Crumley, G., Kaplow, J. M., Schlessinger, J., Jaye, M. and Dionne,C. A. (1991). Ligand-induced transphosphorylation between different FGFreceptors. EMBO J. 10, 2849-2854.
Berghella, L., de Angelis, L., Coletta, M., Berarducci, B., Sonnino, C.,Salvatori, G., Anthonissen, C., Cooper, R., Butler-Browne, G. S., Mouly, V.et al. (1999). Reversible immortalization of human myogenic cells by site-specific excision of a retrovirally transferred oncogene. Hum. Gene Ther. 10,1607-1617.
Böttcher, R. T. and Niehrs, C. (2005). Fibroblast growth factor signalling duringearly vertebrate development. Endocr. Rev. 26, 63-77.
Chen, W. V. and Soriano, P. (2003). Gene trap mutagenesis in embryonic stemcells. Methods Enzymol. 365, 367-386.
Ciruna, B. and Rossant, J. (2001). FGF signaling regulates mesoderm cell fatespecification and morphogenetic movement at the primitive streak. Dev. Cell 1,37-49.
Ciruna, B., Schwartz, L., Harpal, K., Yamaguchi, T. P. and Rossant, J. (1997).Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: a rolefor FGFR1 in morphogenetic movement through the primitive streak.Development 124, 2829-2841.
Copp, A. J. (1985). Relationship between timing of posterior neuropore closureand development of spinal neural tube defects in mutant (curly tail) and nurmalmouse embryos in culture. J. Embryol. Exp. Morphol. 88, 39-54.
Copp, A. J., Seller, M. J. and Polani, P. E. (1982). Neural tube development inmutant (curly tail) and normal mouse embryos: the timing of posteriorneuropore closure in vivo and in vitro. J. Embryol. Exp. Morphol. 69, 151-156.
Copp, A. J., Greene, N. D. E. and Murdoch, J. N. (2003). The genetic basis ofmammalian neurulation. Nat. Rev. Genet. 4, 784-793.
Corson, L. B., Yamanaka, Y., Lai, K. M. and Rossant, J. (2003). Spatial andtemporal patterns of ERK signaling during mouse embryogenesis. Development130, 4527-4537.
Cross, M. J., Lu, L., Magnusson, P., Nyqvist, D., Holmqvist, K., Welsh, M. andClaesson-Welsh, L. (2002). The Shb adaptor protein binds to tyrosine 766 inthe FGFR-1 and regulates the Ras/MEK/MAPK pathway via FRS2 phosphorylationin endothelial cells. Mol. Biol. Cell 13, 2881-2893.
Deng, C.-X., Wynshaw-Boris, A., Shen, M. M., Daugherty, C., Ornitz, D. M.and Leder, P. (1994). Murine FGFR-1 is required for early postimplantationgrowth and axial organization. Genes Dev. 8, 3045-3057.
Deng, C., Wynshaw-Boris, A., Zhou, F., Kuo, A. and Leder, P. (1996). Fibroblastgrowth factor receptor 3 is a negative regulator of bone growth. Cell 84, 911-921.
Fambrough, D., McClure, K., Kazlauskas, A. and Lander, E. S. (1999). Diversesignaling pathways activated by growth factor receptors induce broadlyoverlapping, rather than independent, sets of genes. Cell 97, 727-741.
Farley, F. W., Soriano, P., Steffen, L. S. and Dymecki, S. M. (2000). Widespreadrecombinase expression using FLPeR (flipper) mice. Genesis 28, 106-110.
Finnell, R. H., Gould, A. and Spiegelstein, O. (2003). Pathobiology and geneticsof neural tube defects. Epilepsia 44, S14-S23.
Frank, D. U., Fotheringham, L. K., Brewer, J. A., Muglia, L. J., Tristani-Firouzi, M., Capecchi, M. R. and Moon, A. M. (2002). An Fgf8 mousemutant phenocopies human 22q11 deletion syndrome. Development 129,4591-4603.
Gofflot, F., Hall, M. and Morriss-Kay, G. M. (1997). Genetic patterning of thedeveloping mouse tail at the time of posterior neuropore closure. Dev. Dyn. 210,431-445.
Gotoh, N., Laks, S., Nakashima, M., Lax, I. and Schlessinger, J. (2004). FRS2family docking proteins with overlapping roles in activation of MAP kinase have
RESEARCH ARTICLE Development 133 (4)

DEVELO
PMENT
distinct spatial-temporal patterns of expression of their transcripts. FEBS Lett.564, 14-18.
Gotoh, N., Manova, K., Tanaka, S., Murohashi, M., Hadari, Y., Lee, A.,Hamada, Y., Hiroe, T., Ito, M., Kurihara, T. et al. (2005). The docking proteinFRS2alpha is an essential component of multiple fibroblast growth factorresponses during early mouse development. Mol. Cell. Biol. 25, 4105-4116.
Greco, T. L., Takada, S., Newhouse, M. M., McMahon, J. A., McMahon, A. P.and Camper, S. A. (1996). Analysis of the vestigial tail mutation demonstratesthat Wnt-3a gene dosage regulates mouse axial development. Genes Dev. 10,313-324.
Hadari, Y. R., Gotoh, N., Kouhara, H., Lax, I. and Schlessinger, J. (2001).Critical role for the docking-protein FRS2� in FGF receptor-mediated signaltransduction pathways. Proc. Natl. Acad. Sci. USA 98, 8578-8583.
Hamilton, T. G., Klinghoffer, R. A., Corrin, P. D. and Soriano, P. (2003).Evolutionary divergence of platelet-derived growth factor alpha receptorsignaling mechanisms. Mol. Cell. Biol. 23, 4013-4025.
Hébert, J. M., Lin, M., Partanen, J., Rossant, J. and McConnell, S. K. (2003).FGF signaling through FGFR1 is required for olfactory bulb morphogenesis.Development 130, 1101-1111.
Johnson, D. E. and Williams, L. T. (1993). Structural and functional diversity inthe FGF receptor multigene family. Adv. Cancer Res. 60, 1-41.
Klinghoffer, R. A., Mueting-Nelsen-P. F., Faerman, A., Shani, M. and Soriano,P. (2001). The two PDGFRs display conserved signaling in vivo despite divergentembryological functions. Mol. Cell 7, 343-354.
Klinghoffer, R. A., Hamilton, T. G., Hoch, R. and Soriano, P. (2002). An allelicseries at the PDGF�R locus indicates unequal contributions of distinct signalingpathways during development. Dev. Cell 2, 103-113.
Klint, P. and Claesson-Welsh, L. (1999). Signal transduction by fibroblast growthfactor receptors. Front. Biosci. 4, D165-D177.
Kouhara, H., Hadari, Y. R., Spivak-Kroizman, T., Schilling, J., Bar-Sagi, D.,Lax, I. and Schlessinger, J. (1997). A lipid-anchored Grb2-binding protein thatlinks FGF-receptor activation to the Ras/MAPK pathway. Cell 89, 693-702.
Kovalenko, D., Yang, X., Nadeau, R. J., Harkins, L. K. and Friesel, R. (2003).Sef inhibits fibroblast growth factor signaling by inhibiting FGFR1 tyrosinephosphorylation and subsequent ERK activation. J. Biol. Chem. 278, 14087-14091.
Kuhlbrodt, K., Herbarth, B., Sock, E., Hermans-Borgmeyer, I. and Wegner,M. (1998). Sox10, a novel transcriptional modulator in glial cells. J. Neurosci. 18,237-250.
Larrson, H., Klint, P., Landgren, E. and Claesson-Welsh, L. (1999). Fibroblastgrowth factor receptor-1 mediated endothelial cell proliferation is dependent onthe src homology (SH2/SH3) domain-containing adaptor protein Crk. J. Biol.Chem. 274, 25726-25734.
Macatee, T. L., Hammond, B. P., Arenkiel, B. R., Francis, L., Frank, D. U. andMoon, A. M. (2003). Ablation of specific expression domains reveals discretefunctions of ectoderm- and endoderm-derived FGF8 during cardiovascular andpharyngeal development. Development 130, 6361-6374.
Madhani, H. D. (2001). Accounting for specificity in receptor tyrosine kinasesignaling. Cell 106, 9-11.
Maina, F., Pante, G., Helmbacher, F., Andres, R., Porthin, A., Davies, A. M.,Ponzetto, C. and Klein, R. (2001). Coupling Met to specific pathways results indistinct developmental outcomes. Mol. Cell 7, 1293-1306.
McDougall, K., Kubu, C., Verdi, J. M. and Meakin, S. O. (2001). Developmentalexpression patterns of the signaling adapters FRS-2 and FRS-3 during earlyembryogenesis. Mech. Dev. 103, 145-148.
Mohammadi, M., Honegger, A. M., Rotin, D., Fischer, R., Bellot, F., Li, W.,Dionne, C. A., Jaye, M., Rubinstein, M. and Schlessinger, J. (1991). Atyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factorreceptor (Flg) is a binding site for the SH2 domain of phospholipase C-�1. Mol.Cell. Biol. 11, 5068-5078.
Ong, S. H., Goh, K. C., Lim, Y. P., Low, B. C., Klint, P., Claesson-Welsh, L.,Cao, X., Tan, Y. H. and Guy, G. R. (1996). SUC1-associated neurotrophicfactor (SNT) protein is a major FGF-stimulated tyrosine phosphorylated 90-kDaprotein which binds to the SH2 domain of GRB2. Biochem. Biophys. Res.Commun. 225, 1021-1026.
Ong, S. H., Guy, G. R., Hadari, Y. R., Laks, S., Gotoh, N., Schlessinger, J. andLax, I. (2000). FRS2 proteins recruit intracellular signaling pathways by bindingto diverse targets on fibroblast growth factor and nerve growth factor receptors.Mol. Cell. Biol. 20, 979-989.
Ornitz, D. M. (2000). FGFs, heparan sulfate and FGFRs: complex interactionsessential for development. BioEssays 22, 108-112.
Orr-Urtreger, A., Givol, D., Yayon, A., Yarden, Y. and Lonai, P. (1991).Developmental expression of two murine fibroblast growth factor receptors, flgand bek. Development 113, 1419-1434.
Partanen, J., Schwartz, L. and Rossant, J. (1998). Opposite phenotypes ofhypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1in anteroposterior patterning of mouse embryos. Genes Dev. 12, 2332-2344.
Passos-Bueno, M. R., Wilcox, W. R., Jabs, E. W., Sertié, A. L., Alonso, L. G.and Kitoh, H. (1999). Clinical spectrum of fibroblast growth factor receptormutations. Hum. Mutat.14, 115-125.
Peeters, M. C. E., Schutte, B., Lenders, M.-H. J. N., Hekking, J. W. M.,Drukker, J. and van Straaten, H. W. M. (1998). Role of differential cellproliferation in the tail bud in aberrant mouse neurulation. Dev. Dyn. 211, 382-389.
Pogulis, R. J., Vallejo, A. N. and Pease, L. R. (2000). Recombination andmutagenesis by overlap extension PCR. In The Nucleic Acid Protocols Handbook.(ed. R. Rapley), pp. 857-873. Totowa (NJ): Humana Press.
Rabin, S. J., Cleghon, V. and Kaplan, D. R. (1993). SNT1 a differentiation-specific target of neurotrophic factor-induced tyrosine kinase activity in neuronsand PC12 cells. Mol. Cell. Biol. 13, 2203-2213.
Reilly, J. F., Mickey, G. and Maher, P. A. (2000). Association of fibroblast growthfactor receptor 1 with the adaptor protein Grb14. J. Biol. Chem. 275, 7771-7778.
Roelink, H. and Nusse, R. (1991). Expression of two members of the Wnt familyduring mouse development – restricted temporal and spatial patterns in thedeveloping neural tube. Genes Dev. 5, 381-388.
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases. Cell 103, 211-225.
Shum, A. S. W. and Copp, A. J. (1996). Regional differences in morphogenesis ofthe neuroepithelium suggest multiple mechanisms of spinal neurulation in themouse. Anat. Embryol. 194, 65-73.
Takada, S., Stark, K. L., Shea, M. J., Vassileva, G., McMahon, J. A. andMcMahon, A. P. (1994). Wnt-3a regulates somite and tailbud formation in themouse embryo. Genes Dev. 8, 174-189.
Tallquist, M. D. and Soriano, P. (2000). Epiblast-restricted Cre expression inMORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function.Genesis 26, 113-115.
Tallquist, M. D., Klinghoffer, R. A., Heuchel, R., Mueting-Nelsen, P. F., Corrin,P. D., Heldin, C.-H., Johnson, R. J. and Soriano, P. (2000). Retention ofPDGFR� function in mice in the absence of phosphatidylinositol-3�kinase andphospholipase C� signaling pathways. Genes Dev. 14, 3179-3190.
Tallquist, M. D., French, W. J. and Soriano, P. (2003). Additive effects of PDGFreceptor b signaling pathways in vascular smooth muscle cell development.PLOS Biol.1, 288-299.
Trokovic, N., Trokovic, R., Mai, P. and Partanen, J. (2003a). FGFR1 regulatespatterning of the pharyngeal region. Genes Dev. 17, 141-153.
Trokovic, R., Trokovic, N., Hernesniemi, S., Pirvola, U., Vogt Weisenhorn,D. M., Rossant, J., McMahon, A. P., Wurst, W. and Partanen, J. (2003b).FGFR1 is independently required in both developing mid- and hindbrain forsustained response to isthmic signals. EMBO J. 22, 1811-1823.
Tropepe, V., Siblia, M., Ciruna, B. G., Rossant, J., Wagner, E. F. and van derKooy, D. (1999). Distinct neural stem cells proliferate in response to EGF andFGF in the developing mouse telencephalon. Dev. Biol. 208, 166-188.
Tsang, M., Friesel, R., Kudoh, T. and Dawid, I. B. (2002). Identification of Sef, anovel modulator of FGF signalling. Nat. Cell Biol. 4, 165-169.
Valius, M., Bazenet, C. and Kauzlauskas, A. (1993). Tyrosines 1021 and 1009are phosphorylation sites in the carboxy terminus of the platelet-derived growthfactor receptor beta subunit and are required for binding of phospholipase Cgand a 64-kilodalton protein, respectively. Mol. Cell. Biol. 13, 133-143.
Walshe, J. and Mason, I. (2000). Expression of FGFR1, FGFR2, and FGFR3 duringearly neural development in the chick embryo. Mech. Dev. 90, 103-110.
Wang, J.-K., Xu, H., Li, H.-C. and Goldfarb, M. (1996). Broadly expressed SNT-like proteins link FGF receptor stimulation to activators of the Ras pathway.Oncogene 13, 721-729.
Werner, S., Duan, D.-S. R., de Vries, C., Peters, K. G., Johnson, D. E. andWilliams, L. T. (1992). Differential splicing in the extracellular region ofFibroblast Growth Factor Receptor 1 generates receptor variants with differentligand-binding specificities. Mol. Cell. Biol. 12, 82-88.
Xu, H., Lee, K. W. and Goldfarb, M. (1998). Novel recognition motif onfibroblast growth factor receptor mediates direct association and activation ofSNT adapter proteins. J. Biol. Chem. 273, 17987-17990.
Xu, X., Li, C., Takahashi, K., Slavkin, H. C., Shum, L. and Deng, C.-X. (1999).Murine fibroblast growth factor receptor 1� isoforms mediate node regressionand are essential for posterior mesoderm development. Dev. Biol. 208, 293-306.
Yamaguchi, T. P., Conlon, R. A. and Rossant, J. (1992). Expression of thefibroblast growth factor receptor FGFR-1/flg during gastrulation andsegmentation in the mouse embryo. Dev. Biol. 152, 75-88.
Yamaguchi, T. P., Harpal, K., Henkemeyer, M. and Rossant, J. (1994). fgfr-1 isrequired for embryonic growth and mesodermal patterning during mousegastrulation. Genes Dev. 8, 3032-3044.
Yan, G., Wang, F., Fukabori, Y., Sussman, D., Hou, J. and McKeehan, W. L.(1992). Expression and transforming activity of a variant of the heparin-bindingfibroblast growth factor receptor (flg) gene resulting from splicing of the alphaexon at an alternate 3�-acceptor site. Biochem. Biophys. Res. Comm. 183, 423-430.
Yu, C., Wang, F., Kan, M., Jin, C., Jones, R. B., Weinstein, M., Deng, C.-X. andMcKeehan, W. L. (2000). Elevated cholesterol metabolism and bile acidsynthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J. Biol.Chem. 275, 15482-15489.
673RESEARCH ARTICLEFgfr1-Frs signaling during mouse development