deletion of chromosome ip loci and microsatellite...
TRANSCRIPT

[CANCERRESEARCH55. 5681-5686. December 1. 19951
situ hybridization technique (7, 8), and by detection of LOH3 (9—11).The deletion of lp often involves a large proportion of the distalregion of the short arm, but some tumors and cell lines display smallerdeletions, involving only region 1p36.i—2(9, 10, 12—15).This region,therefore, is a candidate position for one or several NBS gene(s).Further support for a NBS gene in lp36 is given by two cases in whichpatients with neuroblastoma carried constitutional aberrations in thelp36 region (16, 17). Furthermore, in a recent series of articles(13—15,18), several groups presented data suggesting two or moreneuroblastoma tumor suppressor genes in the lp region.
We report that in our patient material, molecular data for lpdeletions, microsatellite instability, and N-myc amplification correlatewith clinical features such as age and stage at presentation andoutcome. Furthermore, a subgroup of neuroblastomas was found tocarry very distal lp deletions. The ip break points in these tumorswere fine mapped using the PCR-based markers, and a proximal anddistal border for a NBS gene could be determined subsequently.
MATERIALS AND METHODS
Patient Material, DNA Extraction, and Southern Analysis Tumor specimens and corresponding normal tissues (fibroblast biopsy or blood sample)were obtained from S1 children, 46 with neuroblastoma of all different stagesand S with ganglioneuroma (Table 1). The clinical material is part of anongoing, population-based Swedish neuroblastoma study. Thus, some of thepatients have been described earlier, and the numbering is in accordance withthat of the earlier report (19). The children were diagnosed, staged, andevaluated for clinical outcome according to the International NeuroblastomaStaging System criteria (20, 21). The 25 children currently surviving have been
followed for 3—92months (median, 32 months), and the remaining 21 diedafter 0—96months (median, 10months; Table 1).Genomic DNA was extractedfrom blood, fibroblasts, and fresh frozen (—70°C)tissue samples using standard procedures. Tumor cell content of the samples was assessed histologicallyin tumor tissue adjacent to that used for DNA extraction. DNA was also
extracted from blood samples from parents of eight patients shown to carry
tumor-specific deletions in the lp region. The N-myc gene copy number wasanalyzed using a Southern blot as reported previously (19). N-myc amplifica
tion was scored when the gene copy number per haploid genome was 3 or
more. Results of N-mycanalyses have been reported previously (19) for someof the tumors included in the current study.
Detection of ip Deletions Using PCR-based DNA Polymorphisms. First,PCR-based polymorphisms located on the distal Ip chromosome region were
analyzed using primer pairs D1580 (22), D1S243, Dl5200 (23, 24), D1S 170,D1S16O (25), and GGAT2AO7 (CHLC, University of Iowa, Iowa City, IA;
Ref. 26). Genetic markers were derived from the CHLC framework map(chromosome 1 framework map, sex-averaged, version 1; via anonymous ftp
to ftp.chlc.org) and for markers D1S8O,D1S170, GGATT2AO7,and MYCLfrom the CHLC likely location map (Likely-bc, version 1). Some neuroblas
tomas found with the first set of markers to carry lp deletions with distal breakpoints were subjected to further study with a more dense marker set derived
from the Genethon 1993—1994map (24). Additional markers used in theseanalyses were D1S468, D1S214, Dl5508, and D1S503.
All primer sequences and PCR conditions were performed according to
published procedures. Primer sequences for GGAT2AO7 were derived via
electronic access from CHLC via anonymous ftp to ftp.chlc.org. The products
3 The abbreviations used are: LOH, loss of heterozygosity; NBS, neuroblastoma tumor
suppressor; CHLC, Cooperative Human Linkage Center; ftp, file transfer protocol.
ABSTRACT
We have analyzed DNA from 46 neuroblastoma tumors of all clinicalstages and five ganglioneuroma tumors together with corresponding control DNA for loss of heterozygosity (L0H) on the distal ip chromosomalregion (lp-LOH). The markers used for the analyses were genetically
mapped DNA polymorphisms detectable with PCR analysis. In general,there was concordance among aggressive tumor stage, ip deletion, andN-myc amplification, although exceptions were found. Twelve (26%) ofthe 46 neuroblastoma tumors displayed lp-LOH, 11 being stage 4 and 1stage 2 (which progressed subsequently to stage 4), whereas 10 stage 4
tumors showed no lp-LOH. Of 12 neuroblastomas shown to have N-mycamplification, 10 had lp-LOH. In 8 cases it was possible to test for
parental origin of the chromosome involved in lp-LOH. No significant
correlation between LOH and paternal or maternal allele was found.Commonly deleted loci in the distal ip region in the neuroblastomatumors indicated that the region for a tentative neuroblastoma tumorsuppressor gene is defined proximally by marker D1S244 and distally by
marker D1SSO. One striking feature of three stage 2 neuroblastomas andone of the stage 3 tumors was the presence in the tumor DNA ofalleles notpresent in the constitutional DNA of the patients, i.e., microsatellite instability. The significance of this phenomenon in localized neuroblastomatumors remains to be clarified.
Aggressive neuroblastoma in young children (younger than 2 years ofage) seems to be a homogenous disorder consistently showing concomitantlp-LOH and N-myc amplification. In the majority of unfavorable neuroblastoma in older children, however, neither lp-LOH nor N-myc amplification could be detected. This indicates that neuroblastoma in olderchildren is a biologically more heterogenous disorder in which geneticalterations other than deletions of chromosome ip and amplification ofN-myc also may contribute to tumongenesis.
INTRODUCTION
Neuroblastoma is a neural crest-derived malignancy, usually occurring during infancy and early childhood. It is the most common
extracranial solid tumor of early childhood, with an annual incidenceof 27.75/106 (about 1/6000) for children younger than 5 years of age(1). Neuroblastoma tumors show marked differences in clinical andbiological behavior, ranging from total spontaneous regression tomalignant progression, giving tumors resistant to therapy. Cytogeneticanalyses of neuroblastoma tumors and cell lines have revealed severalspecific alterations. One of these is the presence of cytogenetic signsof gene amplification, i.e. , double minutes or homogeneously stainingregions. These have been shown to contain amplified copies of theoncogene N-myc (2, 3).
Another alteration present in about 30% of neuroblastomas is adeletion or unbalanced translocation resulting in a loss of geneticmaterial from the distal short arm of chromosome 1 (ip). Thesedeletions have been detected by cytogenetic analysis (4—6),by an in
Received 5/30/95; accepted 9/28/95.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordance with18 U.S.C. Section 1734 solely to indicate this fact.
I Supported by the Swedish Cancer Society, the King Gustav V Jubilee Clinic Cancer
Research Foundation, the Swedish Child Cancer Research Fund, and the Assar Gabrielsson Foundation.
2 To whom requests for reprints should be addressed.
5681
Deletion of Chromosome ip Loci and Microsatellite Instability in Neuroblastomas
Analyzed with Short-Tandem Repeat Polymorphisms1
Tommy Martinsson,2 Rose-Marie Sjoberg, Fredrik Hedborg, and Per Kogner
Department of Clinical Genetics, University of Goteborg, East Hospital, S-416 85 Gothenburg IT. M., R-M. S.]; Department of Pathology, University of Uppsala, UniversityHospital. S-751 85 Uppsala (F. H.]; and Department of Pediatrics, Karolinska Institute, Karolinska Hospital, S-171 76 Stockholm (P. K.], Sweden.
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from

+
+
+
Ip DELETIONS AND MICROSATELLITE INSTABILITY IN NEUROBLASTOMAS
x
A
x90A
Iz
x00A
x90A
x0A
x0A
x
x
x0
x0
x
x0
Emu.o
‘s•o‘ooo+.@
.... I. I•+@
@ 0000@ 0 I
S..... i•+@
...... I.+@
I@ I 000 I
. I. I•I
0000' ‘00
000 I
00000000000' ‘00'•1 IS••••+.@
00100' ‘0
S I••IIII..,..., I+.@0 â€I @00 I I
..... ,..+@
a5'C
soCC
@1C
C
.0C
a5'C
.C
C5'
a
.@
CC5'B
0C
were resolved on a 6% polyacrylamide sequencing gel. After drying, thegel was exposed to XAR-5 film (Eastman KOdak Co.) overnight at roomtemperature.
Constitutional DNA and DNA extracted from the tumor of the patient wererun in adjacent lanes, and the pattern was compared. Essentially, four differentpatterns were obtained: (a) LOH, when the patient was heterozygous, and one
of the alleles was missing or very weak in the tumor DNA compared with thesame allele in constitutional DNA; (b) no LOH, when the patient's containstional DNA was heterozygous, and the corresponding tumor DNA was identical to the constitutional DNA; (c) noninformative, when the patient was
homozygous for the polymorphism in the constitutional DNA, and the tumorDNA gave an identical pattern; and (d) microsatellite instability, when PCRpatterns from tumors display fragments of sizes not present in the constitu
tional DNA.Statistical Analysis. Statistical analysis was performed using Fisher's cx
act test for 2 x 2 tables. Survival probability (±SE)was calculated using theproduct limit method of Kaplan and Meier (27) and compared using theMantel-Haenszel log rank test.
Ethical Approval. The present study was approved as a multicenter studyby the ethics committees of Uppsala University, Karolinska Institute, and theUniversity of Gothenburg.
RESULTS
Detection of LOH and Mapping of ip Deletions. Patients withneuroblastoma (n = 46) and ganglioneuroma (n = 5) were subjectedto analysis with seven PCR-based microsatellite short tandem repeatpolymorphisms (D1S243, DlSl6O, D1S244, D1S228, DlSl7O,GGAT2AO7, and D1S200) and one PCR-based variable number oftandem repeats polymorphism (DIS8O).
Constitutional DNA and DNA extracted from the tumor of thepatient were run in adjacent lanes, and the patterns were compared.Representative analyses are shown in Fig. 1. Twelve of the testedneuroblastomas (26%) but none of five ganglioneuromas (P 0.25)displayed LOH in at least one loci (Table 1).
Two tumors (tumors 121 and 189) displayed LOH for distal markers D1S243 and D15160 but not for more proximal markers D1S244,GA'VF2AO7, and D1S200. Case 52 however, had both alleles retainedfor the most distal marker D1S8O but LOH for more proximal markersD1S243, D15244, and D1S17O. These tumors were subjected toanalyses with more markers in the region to characterize their ipdeletion break points further (Figs. 2 and 3). It could be shown that forcases 121 and 189, D1S5O8 displayed LOH, whereas for both tumors,the two constitutional alleles of marker D1S244 were retained (Figs.2 and 3).
Parental Origin of Deleted Alleles. In eight of the patients withlp-LOH, the parents could be included in the study for testing ofwhich chromosome, the maternal or the paternal, was involved in theLOH process. Subsequently, polymorphisms that were informative
and showed LOH for the children were tested for the family. In fourof the cases (cases 55, 95, 121, and 184), the deleted chromosomeregions were derived from the maternal chromosomes, whereas in thefour other cases (cases 155, 163, 169, and 174), the paternally derivedalleles were deleted in the tumor DNA (Fig. 4).
Presence of Microsatellite Instability in LOCalizedNeuroblastoma. Microsatellite instability was detected in 4 of the 46 testedneuroblastoma tumors (Fig. 1 and Table 1) and, notably, in onlynonmetastasizing tumors (stages 2 and 3; P = 0.077). Especially forthe stage 2 tumors, 3 of the 11 tumors displayed microsatelliteinstability for at least one loci. Case 69, with a stage 3 tumor,displayed microsatellite instability for five of the ip loci tested.D1S200, the most proximal of the tested loci, was informative for thepatient but showed no LOH or microsatellite instability (Fig. 1). Toexclude different possibilities of error, e.g. , contamination or extremedegradation of the DNA, as causes of the microsatellite instability
5682
o• ‘•‘.. I
00000 I @Io@@o@o'g@g
00 I@ I 00'
000' ‘010
00 I I 0 I 00
0 I 000000000000 I I0000'O' I
@1@I0@0 +
000 I 00 I 0
‘0' ‘0000
I ‘00 ‘000
00 I@ I 000
I@ I
00000 I
00'OO' ‘0‘••o‘ooo+@
0 I@ I 0@0 I
01 I I ‘004
4400 ‘004
040 I0000
000'OO' I
0000' I I@
I I I 00 I @I
000000 I 0
000000 I 0
00000 I 0'
0' ‘000 ‘0
I 1000000
I00@0 ‘0
00 I@ I
‘00' I I@0
CC
IU
.0a1-
a.@h
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from
‘pDELETIONS AND MICROSATELLITEINSTABILITY IN NEUROBLASTOMAS
D1S243
52 55 69
NTNT NT
LOH LOH ml.
DISI6O
DIS8O45 51 55 65 68 52 69
N TN TN TN TN TN TNT
-WI.
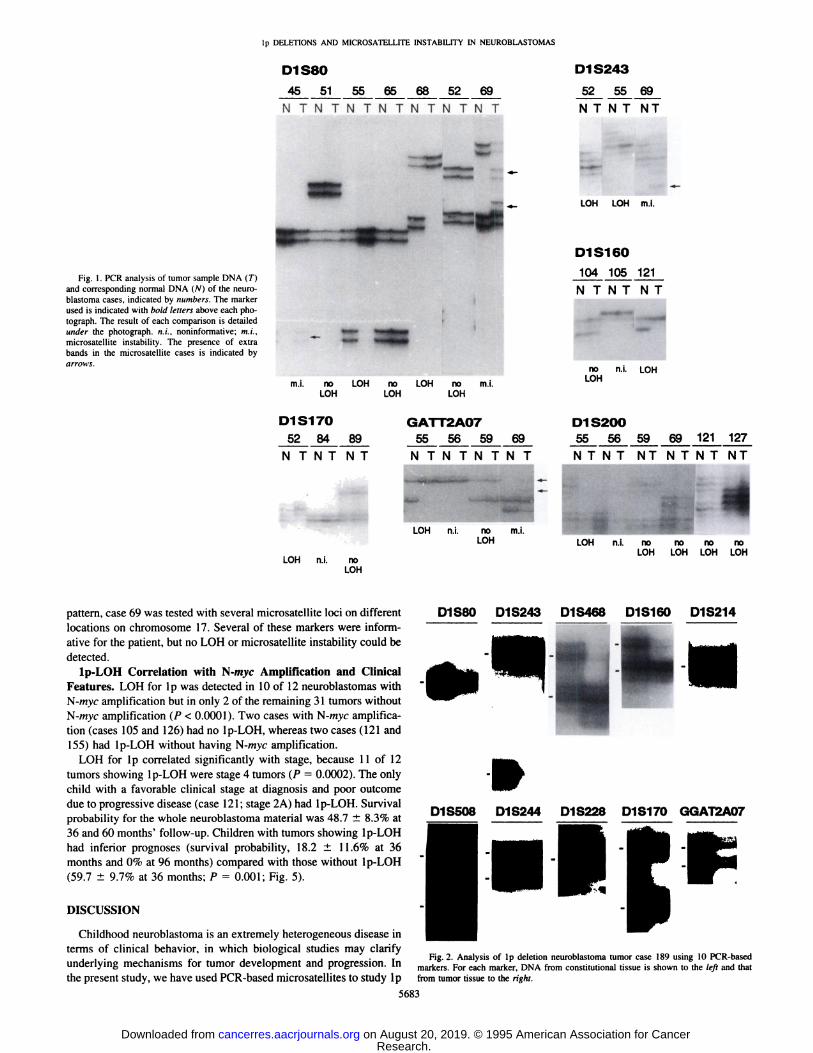
Fig. I . PCR analysis of tumor sample DNA (T) .J9.@... J2@... .1.?!...and corresponding normal DNA (N) of the neuro- N T N T N Tblastoma cases, indicated by numbers. The marker Iused is indicated with bold letters above each photograph. The result of each comparison is detailed @.under the photograph. ni.. noninformative; mi., - ___ Imicrosatellite instability. The pretence of extrabands in the microsatellite cases is indicated byarrows.
no ni. LOH
mi. no LOH no LOH no ml. LOHLOH LOH LOH
DISI7O GATr2AO7 DIS2005284 89 55 56 59 69 55565969121127
NTNTNT NTNTNTNT NTNT NT NTNT NT
@ @- LOH n.i.@
LOH
LOH n.j. noLOH
pattern, case 69 was tested with several microsatellite loci on different DIS8O D1S243 D1S468locations on chromosome 17. Several of these markers were informative for the patient, but no LOH or microsatellite instability could bedetected.
lp-LOH Correlation with N-myc Amplification and ClinicalFeatures. LOH for ip was detected in 10 of 12 neuroblastomas withN-myc amplification but in only 2 of the remaining 31 tumors withoutN-myc amplification (P < 0.0001). Two cases with N-myc amplification (cases 105 and 126) had no lp-LOH, whereas two cases (121 and155) had lp-LOH without having N-myc amplification.
LOH for ip correlated significantly with stage, because 11 of 12tumors showing lp-LOH were stage 4 tumors (P = 0.0002). The onlychild with a favorable clinical stage at diagnosis and poor outcomedue to progressive disease (case 121; stage 2A) had lp-LOH. Survivalprobability for the whole neuroblastoma material was 48.7 ±8.3% at36 and 60 months' follow-up. Children with tumors showing lp-LOHhad inferior prognoses (survival probability, 18.2 ± 11.6% at 36months and 0% at 96 months) compared with those without lp-LOH(59.7 ±9.7% at 36 months; P = 0.001; Fig. 5).
DISCUSSION
Childhood neuroblastoma is an extremely heterogeneous disease interms of clinical behavior, in which biological studies may clarify
. . . Fig. 2. Analysis of lp deletion neuroblastoma tumor case 189 using 10 PCR-based
underlying mechanisms for tumor development and progression. In markers.Foreachmarker,DNAfromconstitutionaltissueis shownto the leftand thatthe present study, we have used PCR-based microsatellites to study ip from tumor tissue to the right.
5683
LOH nI. no@ no r@LOH LOH LOH LOH
D1S1GO D1S214
—I
D1S244 D1S228 DISI7O GGAT2AO7
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from

ip DELETIONS AND MICROSATELLITEINSTABILITY IN NEUROBLASTOMAS
ip
lq
p38.31
p36.13
p36.11
p35.3
p34.2
p33 :
p32.3
p31.3
F—@_1L.D1S2437@
—I—D1S468—018160-
— D1S214
— D1S508
s: D1S244D1S503
10
2
5
9
Hi17
b
37
and found a correlation between ip deletions and N-myc amplification, metastatic phenotype, and an unfavorable clinical outcome.Furthermore, in a limited number of localized tumors, microsatelliteinstability could be detected. From the present investigation, we maysuggest specific clinical subsets with characteristic molecular aberrations.
Using several markers in the distal lp region (Fig. 1), we screeneda group of 5 1 tumors (46 neuroblastomas and 5 ganglioneuromas) forlp-LOH (Table 1 and Fig. 1). LOH for distal lp could be demonstrated in a subset of neuroblastomas (12/46; 26%) but not in any ofthe benign ganglioneuromas. Two cases with distal lp deletions hadtheir deletion break points very distal in ip, i.e., between markersDl 5508 and Dl 5244 (cases 121 and 189; Figs. 2 and 3). The combined information of the 12 cases with lp deletions, therefore, gave aconsensus deletion region for a tentative NBS gene ranging from theproximal marker D1S244 to the distal D1S8O (Fig. 3, bar d). Severalother authors have tried recently to delimit the NBS region by analyzing large numbers of neuroblastoma tumors and defining the commonly deleted regions. White et a!. (28) presented data on a criticaldeleted region defined distally by marker D1Z2 and proximally byD1S228 (Fig. 3, bar a). Caron et a!. (18), however, had no cases ofinterstitial deletions in their material, but the proximal borders of theircommonly deleted region were defined by probe NN1.3 (Fig. 3, barb). This is the GDB locus A12M2, located between markers D1S228and Dl S 199 (18, 26). Amler et a!. (29), in analyzing the commonlydeleted region in a group of neuroblastoma cell lines, defined a criticalregion between markers D1S214 and D1S228 (Fig. 3, bar c). Therefore, a compilation of the earlier published data and those presentedhere indicate a combined region of commonly deleted loci defineddistally by marker D1S214 and proximally by marker D1S244. Thisregion is 7 centimorgans large in the Genethon 1993—1994map (24).
In eight cases, it was possible to determine the parental origin of thelp-LOH chromosome. In four of the cases, the maternal copy was lostin the tumor (Fig. 3), whereas in four tumors, the paternal allele waslost. Caron et a!. (1 1) showed that, in their patient material, thematernal allele was preferentially lost (in 13 of 15 cases), suggestingthat genomic imprinting could be involved in gene regulation in the ipregion. Cheng et al. (30) came to another conclusion. In their study,6 of 10 N-myc-amplified cases involved a deletion of the paternallyderived ip alleles, consistent with a random distribution in theirmaterial. The differences in selection criteria of patients in the twostudies can account possibly for the discrepancy of results. Recently,Caron et a!. (18) adressed the question of imprinting in an extendedstudy of neuroblastoma tumors. Of the 47 tumors with lp-LOHstudied, those with N-myc in single copy had preferentially lost thelp36 allele of maternal origin (16 of 17 cases). These tumors also hada very distal commonly deleted region (i.e., mapping to lp36.3-p36.2). In contrast, all N-myc-amplified neuroblastomas had larger ipdeletions, extending from the telomere to at least lp3S, and thesedeletions were of random parental origin (18 of 30 had maternalLOH). Therefore, Caron et a!. (18) suggested that at least two differ
ent tumor suppressor genes on lp can be inactivated in differentneuroblastoma subtypes. Also, other authors have presented dataindicating more than one suppressor gene in ip (13—15).The data ofour material, although small in number, do contradict at least partlythose of Caron et al. (18). First, case 155 had a lp deletion withoutN-myc amplification, and the deleted copy was of paternal origin.Second, case 189 had the smallest lp deletion in the material butdisplayed N-myc amplification.
One surprising feature of our data was the presence of microsatellite instability in nonmetastasizing neuroblastomas but not in any of
5684
52 121 189
Fig. 3. Delimitation of the commonly deleted region in Ip in neuroblastoma. Data of the lp-LOH study of three neuroblastoma patientt with highly informative Ip deletions. Thetumors were subjected to analysis with a dense set of markers from the Genethon 1993—1994map (24). The origin of the linkage data is described in “Materialsand Methods.―0,no LOH; •,LOH; —,uninformative. The shortest region of overlap of deletions based on the three cases is indicated to the left as bar d. The shortest region of overlap is comparedwith earlier published data of deletions in neuroblastoma tumors and rearrangements in cell lines from White et a!. (28; bar a), Caron et al. (18; bar b), and Amler et a!. (29; bar c).The NBS consensus region of the previous studies and of our present study, defined distally by marker D1S214 and proximally by marker D1S244, is indicated by the gray area.
-9——. . .
— S •
018170
b—GGAT2AO7@ D1S199
@.HMYCL- - -@— D1S200
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from

Ip DELETIONS AND MICROSATELLITE INSTABILITY IN NEUROBLASTOMAS
N-myc amplification without concomitant detection of lp-LOH (13,14, 39). Interestingly, it was reported recently that localized tumorswith N-myc amplification did not necessarily have unfavorable outcomes, especially when a lp deletion was not present (39). A similarfavorable outcome of a stage 4 patient with N-myc amplification butwithout lp-LOH was noted in this material (Table 1, patient 105).Whether the poor outcome for children with tumors showing lp-LOHis due to this aberration alone, or whether the concomitant amplification of N-myc in most cases may be the genetic reason for anaggressive phenotype and poor prognosis, remains to be addressed inlarger studies.
When analyzing our data further, the association of lp-LOH andN-myc amplification was most significant in the children diagnosedwith neuroblastoma younger than the age of 2 years. Six of these hadlp deletions and N-myc amplification, and the other 16 had neitherlp-LOH nor N-myc amplification (P < 0.0001). Among children older
than 2 years of age at diagnosis, 6 showed lp-LOH (four N-mycamplified), and of the remaining 15, 2 had N-myc amplification(P —0.03). All children younger than 2 years of age at diagnosis whodied of tumor progression (5 of 25) had both lp-LOH and N-mycamplification. Among children older than 2 years who died of tumorprogression (15 of 21), 4 had lp-LOH with N-myc amplification, 1had only lp-LOH, 1 had only N-myc amplification, and 9 had neitherlp-LOH nor N-myc amplification. These data indicate clearly that
unfavorable neuroblastoma in older children is a heterogeneous disease by molecular means, whereas in younger children, a deletion ofchromosome lp and a concomitant amplification of N-myc wereconstant findings.
LOH of ip could be detected in a subset of childhood neuroblas
toma tumors in the present material (12 of 46; 26%) almost exclusively in tumors showing unfavorable metastatic stages and poorclinical outcomes. The loss of genetic material confined to this chromosomal region in aggressive tumors indicates the position of one orseveral neuroblastoma tumor suppressor gene(s). Using a panel ofgenetically mapped DNA polymorphisms detectable by PCR, theregion showing LOH in tumor DNA was defined proximally by themarker D1S244 and distally by the marker D1S8O. Microsatelliteinstability could be detected in only nonmetastasizing neuroblastomatumors. The significance of this phenomenon and whether geneticinstability may be of significance in a subset of neuroblastoma tumorsremain to be further investigated. Finally, our data indicated anage-dependent heterogeneity among aggressive neuroblastomas. Inthe present material, a consistent finding was concomitant lp-LOHand N-myc amplification in all unfavorable progressive tumors inchildren younger than 2 years of age. On the other hand, aggressive
5685
121N T MF
1— .
2—@ -@
3—@.@ 3—
1234=1_32=
4—
-@
..o05-
0_100
75
50-@
@Lu11u@@1lM No ip LOH (n—34)
@ II II till 1 II‘.‘
:p=O.OO1-@
.2>@-
(I)L
ip LOH(n=12)25@@,,
:‘‘‘‘‘‘‘
0 12 24 36 48 60 72 8496Months
from diagnosis
Fig. 5. Survival probability according to Kaplan-Meier for 12 children with neuroblastoma with lp-LOH (0%; - - - -) and 34 children with neuroblastoma without detectedlp-LOH (59.7%; —;P = 0.001, Mantel and Hanszel log rank test). Vertical bars indicatechildren alive at last follow-up.
55N T MF
1, 21,2,2 33
1/ 13@1@3 32
@/4@1@ 1@4@1,4 34 4 42
Fig. 4. Analysis of the parental origin of alleles for lp-LOH-detected neuroblastomapatients. Lane N, constitutional DNA of patient; Lane T, patient tumor DNA; Lanes M andF, DNA from mother and father, respectively. Patients 55 and 121 are shown with markerD1S243, and patients 95 and 174 are shown with marker Dl5244. The alleles arenumbered from top to bottom in each family. The genotype of each DNA is indicatedbelow each lane. Tumors of patients 55, 95, and 121 showed loss of the maternal alleles,whereas that of patient 174 showed lots of the paternal allele.
the stage 4 tumors. Microsatellite instability (for recent review seeRef. 31) was observed first in colon cancer (32, 33), and it wassuggested further that it was present preferentially in carcinomas andnot in adenomas (34). In hereditary nonpolyposis colon cancer (Lynchsyndrome) the microsatellite instability is caused apparently by mutations in the MSH2 (35) and MLHJ (36) genes. In one study,Gonzales-Zulueta et a!. (37) found microsatellite instability in sixpatients among 200 with bladder cancer. The six patients were amongthose with lower-stage tumors, whereas no microsatellite instabilitycould be detected in higher-stage tumors. This may suggest thatmicrosatellite instability is an early event in bladder cancer and maycontribute to the progression of tumorigenicity in this tumor type. Inthe neuroblastoma tumors studied by us, microsatellite instability wasfound only in localized tumors, especially in stage 2A and 2B tumors(Table 1). In two other recent reports, microsatellite instability wasinvestigated and detected in only a limited number of neuroblastomatumors without any obvious correlation with clinical features (13, 38).The significance of the phenomenon of microsatellite instability inneuroblastomas demands further study. Whether genetic instabilitymay contribute to the neuroblastoma phenotype in a subset of tumorsremains to be clarified.
We could demonstrate significant correlations among lp-LOH, thepresence of N-myc amplification, and an unfavorable metastatic phenotype. There were, however, cases in our clinical material withlp-LOH without N-myc amplification, as well as the reverse. Previousstudies have given conflicting results concerning a correlation between lp-LOH and N-myc amplification. One study by Caron et al.(11) reported N-myc amplification only in tumors with lp-LOH,whereas other studies, including this one, have found tumors with
95 174NTMF NTMF
@—- -..-_@@
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from

lp DELETIONS AND MICROSATELLITh INSTABIIJTY IN NEUROBLASTOMAS
neuroblastoma in older children seems to be a heterogeneic diseasewith all possible combinations of the presence and absence of ipLOH and N-myc amplification. These data indicate the significant
contribution of several molecular aberrations to tumorigenesis indifferent subsets of neuroblastoma tumors in children older than theage of 2 years.
ACKNOWLEDGMENTS
We acknowledge the clinical and scientific support of Drs. Ildiko Marky,Sven PAhiman,and Magnus Nordenskjdld.
REFERENCES
I. Bernstein, M. L., Leclerc, J. M., Bunin, G., Brisson, L., Robison, L., Shuster, J.,Byrne, T., Gregory. D., Hill, G., Dougherty, G., Scriver, C., Lemieux, B., Tuchman,M., and Woods, W. G. A population-based study of neuroblastoma incidence,survival, and mortality in North America. J. Clin. Oncol., 10: 323—329,1992.
2. Schwab, M., Alitalo, K., Klempnauer, K-H., Varmus, H. E., and Bishop, J. M. AmplifiedDNA with limitedhomologyto mvccellularoncogeneis sharedby humanneuroblastomacell lines and a neuroblastoma tumor. Nature (Lond.), 305: 245-248, 1983.
3. Kohl, N. E., Kanda, N., Schreck, R. R., Brunt, G., Latt, S. A., Gilbert. F., and Alt.,F. W. Transposition and amplification of oncogene-related sequences in humanneuroblastomas. Cell, 35: 359—367,1983.
4. Brodeur,G. M., Sekhon,G. S., andGoldstein,M. N. Chromosomalaberrationsinhuman neuroblastomas. Cancer (Phila.), 40: 2256—2263, 1977.
5. Gilbert, F., Balaban, G., Moorhead, P., Bianchi, D., and Schlesinger, H. Abnormalities of chromosome Ip in human neuroblastoma tumors and cell lines. Cancer Genet.Cytogenet.,7:33—42,1982.
6. Christiansen, H., Schestag, J., Christiansen, N. M., Grzeschik, K. H., and Lampert, F.Clinical impact of chromosome I aberrations in neuroblastoma—a metaphase andinterphase cytogenetic study. Genes Chromosomes & Cancer, 5: 141—149,1992.
7. Zink, D., Weith, A., Martinsson, 1., and Schwab, M. Analysis of chromosome bandlpMl alterations by chromosomal in situ suppression hybridization with a microcloneDNA bank. Genes Chromosomes & Cancer, 3: 407—410, 1991.
8. Stock, C., Ambros, I. M., Mann, G., Gadner, H., Amann, G., and Ambros, P. F.Detection of lp36 deletions in paraffin sections of neuroblastoma tissues. GenesChromosomes & Cancer, 6: 1—9,1993.
9. Fong,C., Dracopoli,N. C., White,P. 5., Merrill,P. T., Griffith,R. C., Housman,D. E., and Brodeur, G. M. Loss of heterozygosity for the short arm of chromosomeI in humanneuroblastomas:correlationwithN-mycamplification.Proc.NatI.Acad.Sci. USA, 86: 3753—3757, 1989.
10. Weith, A., Martinsson, T., Cziepluch. C., Bruderlein, S., Amler, L. C., Berthold, F.,and Schwab, M. Neuroblastoma consensus deletion maps to lp36. 1—2.Genes Chromosomes & Cancer, I: 159—166,1989.
I 1. Caron, H., Vansluis, P., Vanhoeve, M., Dekraker, J., Bras, J., Slater, R., Mannens, M.,Voute, P. A., Westerveld, A., and Versteeg, R. Allelic loss of chromosome lp36 inneuroblastoma is of preferential matemal origin and correlates with N-myc amplification. Nat. Genet., 4: 187—190,1993.
12. Martinsson, T., Weith, A., Cziepluch, C., and Schwab, M. Chromosome I deletionsin human neuroblastomas: generation and fine mapping of microclones from the distallp region. Genes Chromosomes & Cancer, 1: 67—78,1989.
13. Schleiermacher, G., Peter, M., Michon, J., Hugot, J. P., Vielh, P., Zucker, J. M.,Magdelenat, H., Thomas, G., and Delattre, 0. Two distinct deleted regions on theshort arm of chromosome 1 in neuroblastoma. Genes Chromosomes & Cancer, 10:275—281,1994.
14. Takeda, 0., Homma, C., Maseki, N., Sakurai, M., Kanda, N., Schwab, M., Nakamura,Y., and Kaneko, Y. There may be two tumor suppressor genes on chromosome armIp closely associated with biologically distinct subtypes of neuroblastoma. GenesChromosomes & Cancer, 10: 30—39,1994.
15. Cheng, N. C., Van Roy, N., Chan, A., Beitsma, M., Westerveld, A., Speleman, F., andVersteeg, R. Deletion mapping in neuroblastoma cell lines suggests two distincttumor suppressor genes in the lp3S—36region, only one of which is associated withN-myc amplification. Oncogene, 10: 291—297,1995.
16. Laureys,G.. Speleman,F.,Opdenakker,G.,Benoit,Y.,andLeroy,J. Constitutionaltranslocation t(l;l7)(p36;q12—2l) in a patient with neuroblastoma. Genes Chromotomes & Cancer, 2: 252—254,1990.
17. Biegel, J. A., White, P. 5., Marshall, H. N., Fujimori, M., Zackai, E. H., Scher, C. D.,Brodeur, G. M., and Emanuel, B. S. Constitutional lp36 deletion in a child withneuroblastoma. Am. J. Hum. Genet., 52: 176—182,1993.
18. Caron, H., Peter, M., van Sluis, P., Speleman, F., de Kraker, J., Laureys, G., Michon,J., Brugieres, L., Voute, P. A., Westerveld, A., Slater, R., Delattre, 0., and Versteeg,R. Evidence for two tumour suppressor Id on chromosomal bands lp35—36involvedin neuroblastoma: one probably imprinted, another associated with N-myc amplification. Hum. Mol. Genet., 4: 535—539,1995.
19. Hedborg, F., Lindgren, P. G., Johanston, I., Kogner, P., Samuelsson, B-O., Bekassy,A. N., Olsen, L., Kreuger, A., and Pâlman,S. N-myc gene amplification in neuroblastoma: a clinical approach using ultrasound guided cutting needle biopsies collected at diagnosis. Med. Pediatr. Oncol., 20: 292—300,1992.
20. Brodeur, G. M., Seeger, R. C., Barret, A., Berthold, F., Castleberry, R. P., D'Angio,G., DeBernardi, B., Evans, A., Favot, M., Freeman, A. I., Haase, G., Hartmann, 0.,Hayes, F. A., Helson, L., Kemshead, L., Lampeit, F., Niane, J., Ohkawa, H., Philippe,T., Pinkerton, C. R., Pritchard, J., Sawada, T., Siegel, S., Smith, E. I., Tsuchida, Y.,and Voute, P. A. International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J. Clin. Oncol., 6: 1874—1881, 1988.
21. Brodeur, G. M., Pritchard, J., Berthold, F., Carlsen, N. L. 1., Castel, V., Castleberry,R. P., Debemardi, B., Evans, A. E., Favrot, M., Hedborg, F., Kaneko, M., Kemshead,J., Lampert, F., Lee, R. E. J., Look, A. T., Pearson, A. D. J., Philip, T., Roald, B.,Sawada, T., Seeger, R. C., Tsuchida, Y., and Vows, P. A. Revisions of the internstional criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin.Oncol., 11: 1466—1477, 1993.
22. Budowle, B., Chakraborty, R., Giusti, A. M., Eisenberg, A. J., and Allen, R. C.Analysis of the VNTR locus D1580 by the PCR followed by high-resolution PAGE.Am. J. Hum. Genet., 48: 137—144,1991.
23. Weissenbach, J., Gyapay, G., Dib, C., Vignal, A., Morissette, J., Millasseau, P.,Vaysseix, G., and Lathrop, M. A 2nd-generation linkage map of the human genome.Nature (Land.), 359: 794—801, 1992.
24. Gyapay, G., Morissette, J., Vignal, A., Dib, C., Fizames, C., Millasseau, P., Marc, S.,Bernardi, G., Lathrop, M., and Weissenbach, J. The 1993—94Genethon humangenetic linkage map. Nat. Genet., 7: 246—339,1994.
25. Engelstein, M., Hudson, T. J., Lane, J. M., Lee, M. K., Leverone, B., Landes, G. M.,Peltonen, L., Weber, J. L., and Dracopoli, N. C. A PCR-based linkage map of humanchromosome I. Genomics, 15: 251—258,1993.
26. Buetow, K. H., Weber, J. L., Ludwigsen, S., Scherpbierheddema, T., Duyk, G. M.,Sheffield, V. C., Wang, Z. Y., and Murray, J. C. Integrated human genome-wide mapsconstructed using the CEPH reference panel. Nat. Genet., 6: 391—393,1994.
27. Kaplan, E. L., and Meier P. Nonparametric estimation from incomplete observations.J. Am.Stat.Assoc.,53: 457—481,1958.
28. White, P. 5., Mans, J. M., Beltinger, C., Sulman, E., Marshall, H. N., Fujimori, M.,Kaufman, B. A., Biegel, J. A., Allen, C., Hilliard, C., Valentine, M. B., Look, A. 1.,Enomoto, H., Sakiyama, S., and Brodeur, G. M. A. region of consistent deletion inneuroblastoma maps within human chromosome lp36.2—36.3. Proc. NatI. Acad. Sci.USA, 92: 5520—5524, 1995.
29. Amler, L. C., Corvi, R., Pram!, C., Savelyeva, L., Lepaslier, D., and Schwab, M. Areciprocal translocation (1 ;15) (36.2 \4) in a neuroblastoma cell line is accompaniedby DNA duplication and may signal the site of a putative tumor suppressor-gene.Oncogene,10:1095—1101,1995.
30. Cheng, J. M., Hiemstra, J. L., Schneider, S. S., Naumova, A., Cheung, N. K. V.,Cohn, S. L., Diller, L., Sapienza, C., and Brodeur, G. M. Preferential amplification ofthe paternal allele of the N-myc gene in human neuroblastomas. Nat. Genet., 4:191—194,1993.
31. Loeb, L. A. Microsatellite instability: marker of a mutator phenotype in cancer.Cancer Ret., 54: 5059—5063,1994.
32. Aaltonen, L. A., Peltomaki, P., Leach, F. S., Sistonen, P., Pylkkanen, L., Mecklin,J. P., Jarvinen, H., Powell, S. M., Jen, J., Hamilton, S. R., Petersen, G. M., Kinzler,K. W., Vogelstein, B., and de la Chapelle, A. Clues to the pathogenesis of familialcolorectal cancer. Science (Wash. DC), 260: 812—816, 1993.
33. Thibodeau, S. N., Bren, G., and Schaid, D. Microsatellite instability in cancer of theproximal colon. Science (Wash. DC), 260: 816—819, 1993.
34. Young, J., Leggett, B., Gustafton, C., Ward, M., Searle, J., Thomas, L., Buttenshaw,R., and Chenevixtrench, G. Genomic instability occurs in colorectal carcinomas butnot in adenomas. Hum. Mutat., 2: 351—354,1993.
35. Peltomaki, P., Aaltonen, L. A., Sistonen, P., Pylkkanen, L., Mecklin, J. P., Jarvinen,H., Green, J. S., Jass, J. R., Weber, J. L., Leach, F. S., Petersen, G. M., Hamilton,S.R.,de laChapelle,A.,andVogelstein,B.Geneticmappingofa locuspredisposingto human colorectal cancer. Science (Wash. DC), 260: 810—812, 1993.
36. Bronner, C. E., Baker, S. M., Morrison, P. T., Warren, G., Smith, L. G., Lescoe,M. K., Kane, M., Earabino, C., Lipford, J., Lindblom, A., Tannergard, P., Bollag,R. J., Godwin, A. R., Ward, D. C., Nordenskjold, M., Fishel, R., Kolodner, R., andLiskay, R. M. Mutation in the DNA mismatch repair gene homologue hMLH1 isassociated with hereditary non-polyposis colon cancer. Nature (Lond.), 368: 258—261, 1994.
37. Gonzalez-Zulueta, M., Ruppert, J. M., Tokino, K., Tsai, Y. C., Spruck, C. H., Miyao,N., Nichols, P. W., Hermann, G. G., Horn, T., Steven, K., Summerhayes, I. C.,Sidransky, D., and Jones, P. A. Microsatellite instability in bladder cancer. CancerRet., 53: 5620—5623, 1993.
38. Gehring, M., Berthold, F., Schwab, M., and Amler, L. Genetic stability of microsatellites in human neuroblastomas. mt. J. Oncol., 4: 1043—1045,1994.
39. Cohn, S. L., Look, A. T., Joshi, V. V., Holbrook, T., Salwen, H., Chagnovich, D.,Chesler, L., Rowe, S. T., Valentine, M. B., Komuro, H., Castleberry, R. P., Bowman,L. C., Rao,P. V., Seeger,R. C., andBrodeur,G. M. Lackof correlationof N-mycgene amplification with prognosis in localized neuroblastoma: a pediatric oncologygroup study. Cancer Ret., 55: 721—726,1995.
5686
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from

1995;55:5681-5686. Cancer Res Tommy Martinsson, Rose-Marie Sjöberg, Fredrik Hedborg, et al. PolymorphismsNeuroblastomas Analyzed with Short-Tandem Repeat Deletion of Chromosome 1p Loci and Microsatellite Instability in
Updated version
http://cancerres.aacrjournals.org/content/55/23/5681
Access the most recent version of this article at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/55/23/5681To request permission to re-use all or part of this article, use this link
Research. on August 20, 2019. © 1995 American Association for Cancercancerres.aacrjournals.org Downloaded from