heme proteins from (rather) chemical point of view...
TRANSCRIPT

Heme Proteins
from (rather) Chemical Point of View
(Inorganic, Organic, Bio-)
Oct. 2-Oct. 29: 14 times + Test
D107: 5 p.m.-7 p.m. : Mon, Tue, Wed
D105: 12 p.m.-2 p.m. : Fri

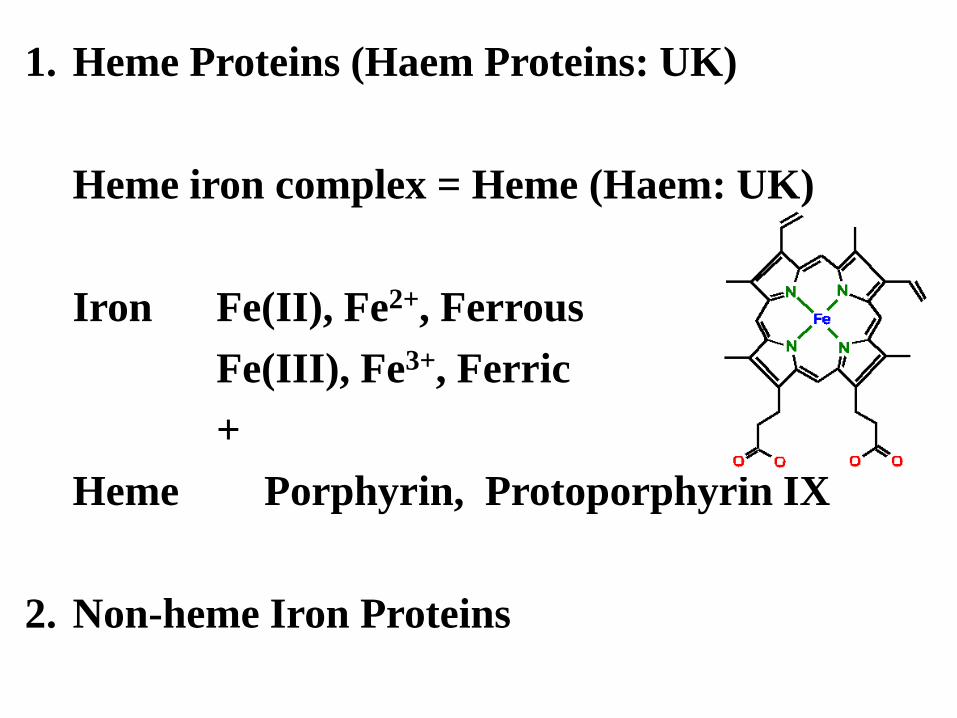
1. Heme Proteins (Haem Proteins: UK)
Heme iron complex = Heme (Haem: UK)
Iron Fe(II), Fe2+, Ferrous
Fe(III), Fe3+, Ferric
+
Heme Porphyrin, Protoporphyrin IX
2. Non-heme Iron Proteins

Figure 2. Chemical structures of representative iron complexes in proteins. Rubredoxin, ferredoxin, [4Fe–4S] clusters, and Rieske [2Fe–2S] clusters are found in proteins that function as electron transfer processes. [2Fe–2S] and [4Fe–4S] clusters are also engaged in regulation of the transcription of genes that are under redox control. Mononuclear iron complexes are found in enzymes that catalyze oxygenation reactions. Oxo-Fe2 complexes catalyze dehydrogenation or reduction of ribonucleotides to deoxyribonucleotides. Fe–Ni hydrogenase and Fe–Fe hydrogenase complexes are found in hydrogenases. The CO dehydrogenase cluster shown is cluster B in carbon monoxide dehydrogenase. The aconitase intermediate occurs as the dehydration intermediate in aconitase. Iron protoporphyin IX is found in heme proteins, cytochromes c and a, and cytochrome P450. The radical SAM initiation complex is found in radical SAM enzymes. FeMoco and P-cluster are essential cofactors in nitrogenase. The interstitial carbide (C4–) has recently been identified.(14, 15) ACS Chem. Biol. 7, 1477 (2012) Radical SAM enzymes use a 4Fe-4S cluster to transfer an electron from an external source
(such as flavodoxin) to an S-adenosylmethionine (SAM) molecule, which converts to methionine and a 5’-deoxyadenosyl radical. This radical is very reactive, and is able to abstract a proton from a C-H that is appropriately placed.


6 6
Cloning, expression and purification
E. coli K-12 genome
cloning
pET21c
overexpression Full-length YddV
97
66
45
(kDa)
54 kDa purification
YddV contains a b-type heme with 1 : 1 stoichiometry.
33

Iron Protoporphyrin IX, heme b Prosthetic group A flat and planar structure. Itself Toxic O2
-., H2O2 are formed.
Heme a Heme a is a form of heme found In cytochromes a and a3.


Heme proteins = heme + proteins
Transfer & storage of gas molecules
O2, NO
Mb, Hb
Oxidation
O2 + e-
P450, NOS
Electron transfer
e-
Cytochrome c
Heme sensor Gas sensor
Heme:HRI O2 , CO, NO: FixL, DOS

Heme Proteins-Agenda but not so strict.
(1) Heme Iron, Iron Homeostasis
(2)Myoglobin, Hemoglobin
(3)Heme Spectra
(4)Cytochrome c, cytochrome b5: Electron transfer
(5)Cytochrome P450 (CYP): O2 activation
(6)Nitric oxide synthase (NOS): O2 activation
(7)NO Sensor: sGC
(1)Heme oxygenase (HO)
(2)Heme Sensor-1: Heme regulated inhibitor (HRI)
(3)Heme Sensor-2: Circadian rhythms
(4)Heme-based oxygen sensor-1: FixL, Ec DOS
(5)Heme-based oxygen sensor-2: YddV
(6)Mammalian oxygen sensor: HIF1a
(7)Heme-based CO sensor, H2S: CooA, RcoM, BK Channel, CBS
(8)Test

Myoglobin O2 Storage
Myoglobin, Hemoglobin: Heme iron complex containing proteins
(Haemoglobin, Haem: UK spelling)

12
You don’t need O2. Less myoglobin.
You need O2 because of continuous swimming. More myoglobin.
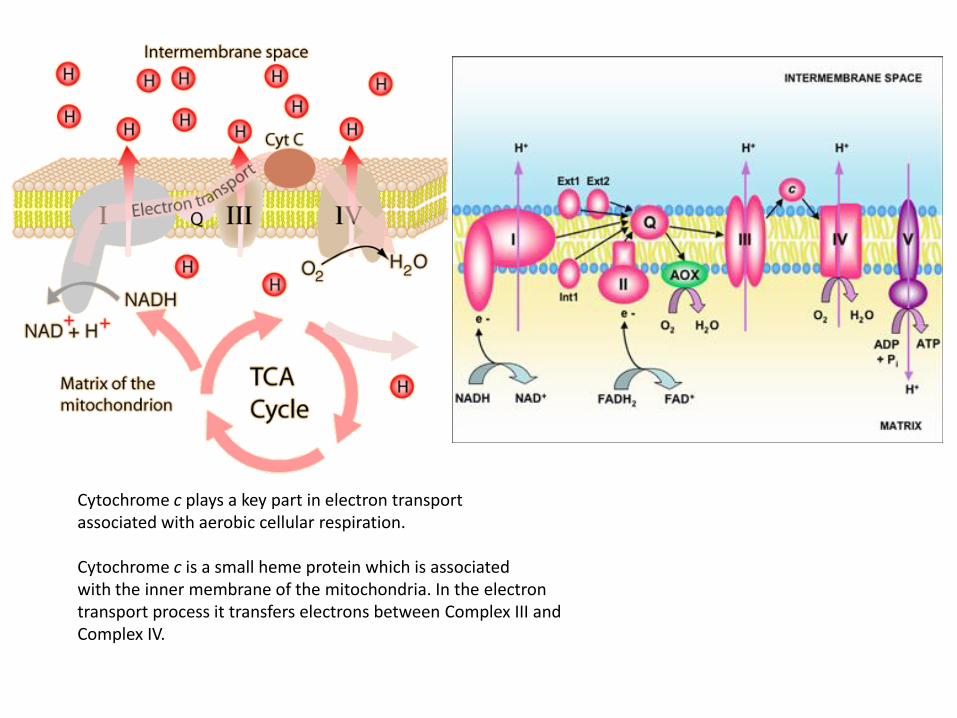
Cytochrome c plays a key part in electron transport associated with aerobic cellular respiration. Cytochrome c is a small heme protein which is associated with the inner membrane of the mitochondria. In the electron transport process it transfers electrons between Complex III and Complex IV.

14
NSAID: non-steroid anti-inflammatory drug

15
Blue Rose: Suntory
Flower color was changed by manipulation of P450 genes: Rose

Heme-assisted S-nitrosation of a proximal thiolate in
a nitric oxide transport protein
Proc. Natl. Acad. Sci. USA 102, 594 (2005)
16 Rhodnius prolixus (the kissing bug)
Cimex lectularius (the bedbug)
Vasorelaxation +
Platelet Inhibition

17
Vasorelaxation
Platelet Inhibition
Soluble guanylate
cyclase (sGC)

Figure 12. Summarized illustration of the main, significant categories of NO reactions in the vascular system. The reactions include (1) reactions with metal centers (mainly heme); (2) S-nitrosylation, or the interaction of NO with cysteine sulfahydryls/thiol, where a nitrosyl group is added post-translationally; (3) nitration (protein tyrosine); (4) free-radical interactions; (5) reactions with plasma O2; and (6) synthesis of cGMP through the catalysis of sGC by NO, then leading to the activation of protein kinases and phosphodiesterases.

19
Sensor domain
(Heme) Functional domain
Protein
structural
change
Sensor domain
(Gas-bound heme)
Functional domain Regulation of
catalysis and
transcription
Signal (O2, CO, NO)
Ec DOS, FixL, HemAT, sGC, CooA
Heme-based gas sensor protein
Heme
Heme

Sensor domain Function domain
Protein
structural
changes
Sensor domain
(heme-bound)
Function domain Catalysis,
DNA binding
Heme-responsive heme-sensor proteins
N N
N N
O
O
O
O
Heme as the
1st signal
Fe
20

Pharm. Rev. 60, 79 (2008)
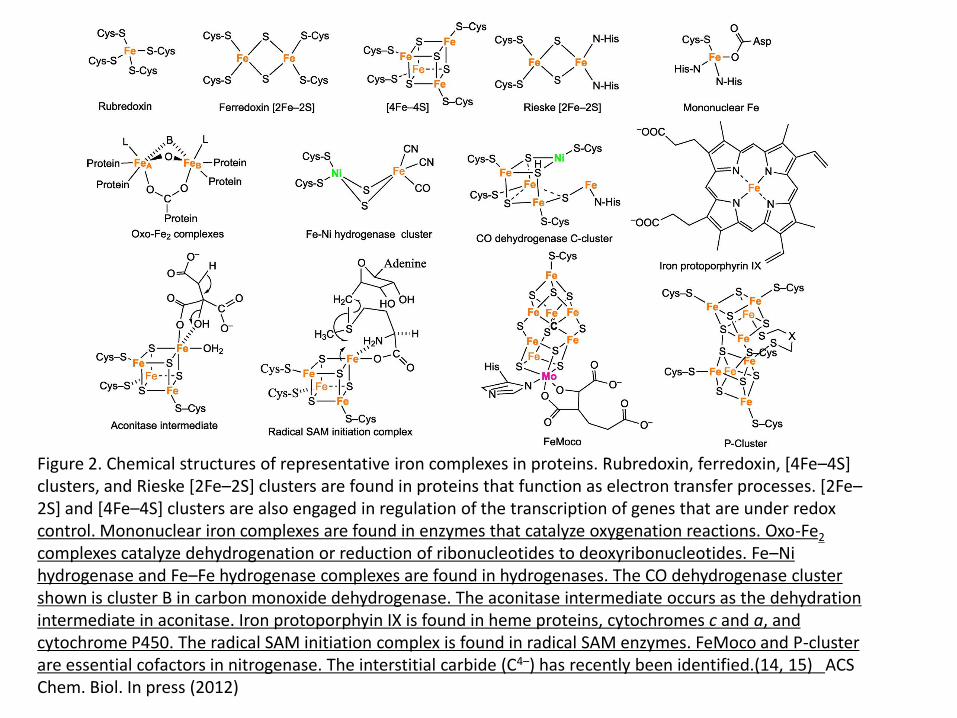
Figure 2. Chemical structures of representative iron complexes in proteins. Rubredoxin, ferredoxin, [4Fe–4S] clusters, and Rieske [2Fe–2S] clusters are found in proteins that function as electron transfer processes. [2Fe–2S] and [4Fe–4S] clusters are also engaged in regulation of the transcription of genes that are under redox control. Mononuclear iron complexes are found in enzymes that catalyze oxygenation reactions. Oxo-Fe2 complexes catalyze dehydrogenation or reduction of ribonucleotides to deoxyribonucleotides. Fe–Ni hydrogenase and Fe–Fe hydrogenase complexes are found in hydrogenases. The CO dehydrogenase cluster shown is cluster B in carbon monoxide dehydrogenase. The aconitase intermediate occurs as the dehydration intermediate in aconitase. Iron protoporphyin IX is found in heme proteins, cytochromes c and a, and cytochrome P450. The radical SAM initiation complex is found in radical SAM enzymes. FeMoco and P-cluster are essential cofactors in nitrogenase. The interstitial carbide (C4–) has recently been identified.(14, 15) ACS Chem. Biol. In press (2012)

Mammalian Mechanisms of Iron Homeostasis
Biochemistry 51, 5705 (2012)
See Table 1, Table 2



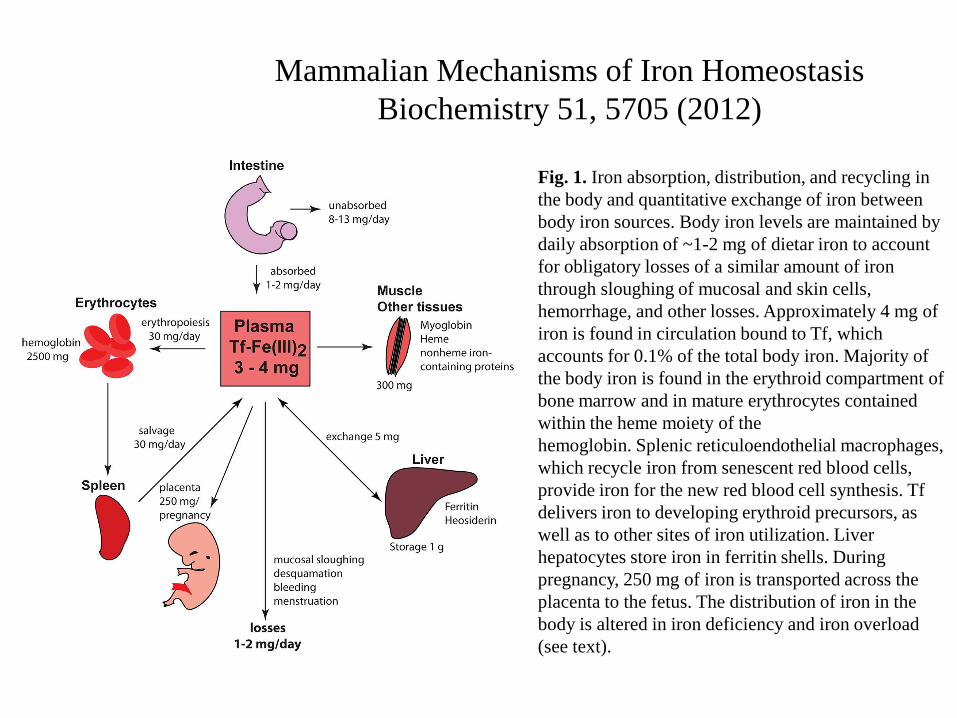
Mammalian Mechanisms of Iron Homeostasis
Biochemistry 51, 5705 (2012)
Fig. 1. Iron absorption, distribution, and recycling in
the body and quantitative exchange of iron between
body iron sources. Body iron levels are maintained by
daily absorption of ~1-2 mg of dietar iron to account
for obligatory losses of a similar amount of iron
through sloughing of mucosal and skin cells,
hemorrhage, and other losses. Approximately 4 mg of
iron is found in circulation bound to Tf, which
accounts for 0.1% of the total body iron. Majority of
the body iron is found in the erythroid compartment of
bone marrow and in mature erythrocytes contained
within the heme moiety of the
hemoglobin. Splenic reticuloendothelial macrophages,
which recycle iron from senescent red blood cells,
provide iron for the new red blood cell synthesis. Tf
delivers iron to developing erythroid precursors, as
well as to other sites of iron utilization. Liver
hepatocytes store iron in ferritin shells. During
pregnancy, 250 mg of iron is transported across the
placenta to the fetus. The distribution of iron in the
body is altered in iron deficiency and iron overload
(see text).

Fig. 2. Regulation of systemic iron
metabolism. Organs and cell types
involved in systemic iron balance are
shown. Duodenal enterocytes absorb
dietary iron via DMT1 located on the
apical surface upon reduction of Fe3+ to
Fe2+ by DcytB. Spleenic
reticuloendothelial macrophages recycle
iron from senescent red blood cells.
Both cell types release iron via
ferroportin with the aid of hephaestin,
which oxidizes Fe2+ to Fe3+. Iron is
also oxidized by ceruloplasmin in the
circulation. Plasma Tf captures and
circulates iron in the body. Hepatic
hormone, hepcidin regulates iron efflux
from these cells by regulating the
stability of ferroportin. Synthesis and
secretion of hepcidin by hepatocytes is
influenced
by iron levels in the body as well as
conditions that affect iron metabolism
indirectly such as
inflammation, ER stress, erythropoiesis,
and hypoxia (see text for additional
details).

Fig. 3. Cellular iron metabolism. Most
cells in the body obtain iron from
circulating differic Tf. Iron loaded holo-
Tf binds to TfR1 on the cell surface and
the complex undergoes endocytosis via
clathrin coated pits. A proton pump
acidifies the endosome resulting in the
release of Fe3+, which is subsequently
reduced to Fe2+ by Steap3 and
transported across the endosomal
membrane to the cytosol by DMT1.
DMT1 also facilitates dietary iron
absorption. Apo-Tf is recycled back to the
cell surface and released from TfR1 to
plasma to repeat another cycle. Newly
acquired iron enters into cytosolic “labile
iron pool” (LIP), which is redox-active.
LIP is chelated by intracellular
siderophore that facilitates intracellular
iron trafficking to mitochondria via an
unknown receptor for metabolic
utilization (such as synthesis of heme and
iron-sulfur clusters), and cellular iron that
is not utilized is either stored in ferritin or
exported via ferroportin. Cells also export
iron contained in ferritin and heme.

Fig. 4. Cellular iron balance. A typical IRE motif consists of a hexanucleotide loop with the sequence 5′-CAGUGH-3′ (H
could be A, C, or U) and a stem, interrupted by a bulge with an unpaired C residue. IREs post-transcriptional control
expression of regulators of cellular iron metabolism in concert with IRPs. Translational-type IRE/IRP interactions in the 5’
UTR modulate the expression of the mRNAs encoding H- and L-ferritin, ALAS2, m-aconitase, ferroportin, and HIF-2α,
which in turn control iron storage, erythroid iron utilization, energy homeostasis, iron efflux, and hypoxic responses,
respectively. Conversely, IRE/IRP interactions in the 3’ UTR stabilize the mRNAs encoding TfR1, DMT1, and Cdc14A,
which are involved in iron uptake, iron transport, and the cell cycle, respectively. Under physiological conditions, IRP1 is
regulated by a reversible ISC switch. Iron deficiency, promotes ISC disassembly and a conformational rearrangement,
resulting in conversion of IRP1 from c-aconitase to an IRE-binding protein. The ISC is regenerated in iron-replete cells.
Hypoxia favors maintenance of the ISC, while H2O2 promotes its disassembly. When the ISC biogenesis pathway is not
operational, iron leads to ubiquitination of apo-IRP1 by the FBXL5 E3 ligase complex (including Skp1, Cul1 and Rbx1),
resulting in proteasomal degradation. IRP2 is stable in iron deficient and/or hypoxic cells; under these conditions FBXL5
undergoes ubiquitination and proteasomal degradation. An increase in iron and oxygen levels stabilizes FBXL5 by
formation of an Fe-O-Fe center in its hemerythrin domain, triggering the assembly of an E3 ubiquitin ligase complex
together with Skp1, Cul1 and Rbx1. This complex ubiquitinates IRP2, leading to its recognition by the proteasome and its
degradation.

Supplementation With Oral vs. Intravenous Iron for Anemia With IBD (inflammatory bowel disease) or Gastrointestinal Bleeding: Is Oral Iron Getting a Bad Rap? Amer. J. Gastroenterology 106, 1872 (2011) Ferric iron (Fe3+) is reduced to ferrous iron (Fe2+) in the intestinal lumen by ferrireductase. Ferrous iron (Fe2+) is transported across the enterocyte by divalent metal ion transporter-1 (DMT-1). Dietary heme iron is transported across the enterocyte by heme carrier protein 1 (HCP1). Inside the enterocyte, heme is acted upon by heme oxygenase, which releases ferrous iron from the protoporphyrin in heme. The released iron enters the same pool as dietary non-heme iron. A portion of this pool is stored as ferritin inside the enterocyte, which is later lost with sloughing of the intestinal mucosa. The remainder of the ferrous iron is transported across the basolateral membrane of the enterocyte by ferroportin. The transported Fe2+ is oxidized back to the Fe3+ form by ferroxidase. Fe3+ is then incorporated into transferrin in the serum. The protein hepcidin regulates iron transport into serum by causing internalization of ferroportin with subsequent lysosomal breakdown inside the enterocyte.

Cell Metabolism 16, 449 (2012)
Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic
b Cell Fate in Response to Cytokines
Reactive oxygen species (ROS) contribute to target-cell damage in
inflammatory and iron-overload diseases. Little is known about iron
transport regulation during inflammatory attack. Through a combination of
in vitro and in vivo studies, we show that the proinflammatory cytokine IL-
1b induces divalent metal transporter (DMT1) expression correlating with
increased b cell iron content and ROS production. Iron chelation and
siRNA and genetic knockout of DMT1 expression reduce cytokine-induced
ROS formation and cell death. Glucose-stimulated insulin secretion in the
absence of cytokines in Dmt1 knockout islates is defective, highlighitng a
physiological role of iron and ROS in the regulation of insulin secretion.
Dmt1 knockout mice are protected against multiple low-doze
streptozotoucin and high-fat diet-induced glucose intolerance, models of
type 1 and type 2 diabetes, respectively. Thus, b cells become prone to
ROS-mediated inflammatory damage via aberrant cellular iron metabolism,
a finding with potential general cellular implications.


Human Iron−Sulfur Cluster Assembly, Cellular Iron Homeostasis, and Disease Biochemistry Current Topics 49 (24) 4945 (2010)
Iron−sulfur (Fe−S) proteins contain prosthetic groups consisting of two or more iron atoms bridged by sulfur ligands,
which facilitate multiple functions, including redox activity, enzymatic function, and maintenance of structural integrity.
More than 20 proteins are involved in the biosynthesis of iron−sulfur clusters in eukaryotes. Defective Fe−S cluster
synthesis not only affects activities of many iron−sulfur enzymes, such as aconitase and succinate dehydrogenase, but
also alters the regulation of cellular iron homeostasis, causing both mitochondrial iron overload and cytosolic iron
deficiency. In this work, we review human Fe−S cluster biogenesis and human diseases that are caused by defective
Fe−S cluster biogenesis. Fe−S cluster biogenesis takes place essentially in every tissue of humans, and products of
human disease genes, including frataxin, GLRX5, ISCU, and ABCB7, have important roles in the process. However,
the human diseases, Friedreich ataxia, glutaredoxin 5-deficient sideroblastic anemia, ISCU myopathy, and ABCB7
sideroblastic anemia/ataxia syndrome, affect specific tissues, while sparing others. Here we discuss the phenotypes
caused by mutations in these different disease genes, and we compare the underlying pathophysiology and discuss the
possible explanations for tissue-specific pathology in these diseases caused by defective Fe−S cluster biogenesis.

Figure 1. Subcellular iron trafficking in human cells. Iron is bound to transferrin Tf in serum, which interacts with
TfR1 on the cell membrane. Formation of the Tf−TfR1 complex induces endocytosis. Upon acidification of the
endosome, iron is released and then exported into the cytosol by DMT1 or TRPML1. Iron in cytosol has four main
fates. It can be used to assemble Fe−S clusters and other iron proteins in cytosol, undergo transport into
mitochondria via the mitochondrial iron importer, mitoferrin, be loaded into ferritin with the coordination of the
PCBP1 chaperone, or be exported by ferroportin. Upon its import into mitochondria, iron is used to synthesize
Fe−S clusters and heme. An unknown compound that represents the mitochondrial iron status and is a product of
Fe−S cluster synthesis appears to be exported by ABCB7, which can affect cytosolic Fe−S cluster biogenesis.
Heme is exported from the mitochondria by an unknown mechanism and is exported out of some cells by a heme
transporter known as FLVCR.


Figure 2. Comparison of Fe−S cluster biogenesis pathways in eukaryotes. In yeast mitochondria, Nfs1 and Isd11
form a complex of cysteine desulfurase, which provides sulfur to scaffold proteins for Fe−S cluster assembly.
The pair of ferredoxin (Yah1) and ferredoxin reductase (Arh1) perhaps provides reducing equivalents needed for
cluster assembly. The yeast frataxin homologue, Yfh1, provides iron. Iron−sulfur clusters are assembled on
scaffolds, and there are several alternative scaffolds, including Isu1 and -2, Nfu, Isa1 and -2, and Grx5. Iba57 is
thought to function with the Isa1 and -2 scaffold proteins. Facilitated by chaperone (Ssq1) and cochaperone
(Jac1) activities, the preassembled Fe−S clusters of Isu1 and -2 are delivered to target apoproteins in
mitochondria. The mitochondrial inner membrane transporter Atm1 has been proposed to export either Fe−S
cluster or sulfur to the cytosol, with assistance from Erv1 in the intermembrane space of mitochondria, and this
compound “X” could then be used to assemble Fe−S clusters in the cytosol by scaffold proteins, including Cfd1,
Nbp35, Nar1, and Cia1. The assembled Fe−S clusters are delivered to apoproteins, including the Grx3/4−Fra2
complex, which regulates the nucleocytoplasmic translocation of Aft1 by an unknown mechanism. As a
transcription factor, Aft1 regulates transcription of the iron regulon in the yeast nucleus. Interestingly, there is a
fraction of Nfs1 proteins present in yeast nucleus (109). In human mitochondria, ISCS and ISD11 form a
complex that provides sulfur to scaffold proteins for Fe−S cluster biogenesis, while FXN is thought to provide
iron. Potential scaffold proteins include ISCU, NFU, ISCA1/2, and GLRX5. The assembled Fe−S clusters are
delivered to apoproteins for maturation. The roles of human ferredoxin, ferredoxin reductase, Iba57, chaperone,
and cochaperone homologues in mitochondrial Fe−S cluster biogenesis remain to be confirmed. An unknown
molecule that depends on mitochondrial Fe−S cluster biogenesis for function is exported by ABCB7. We propose
that the ABCB7-exported molecule may serve as a signal that induces iron transcriptional remodeling in the
nucleus to appropriately regulate mitochondrial iron homeostasis in response to a signal received from the
mitochondria. In the cytosol, the cytosolic forms of ISCS and ISD11, c-ISCS and c-ISD11, respectively, provide
sulfur, while iron can be directly acquired from cytosol, though its exact molecular source is not known. Fe−S
clusters are likely synthesized de novo on c-ISCU, c-NFU, c-ISCA1, or NUBP1 (Nbp35) scaffolds. IOP1 may
function in the delivery of the cluster to apoproteins. The involvement of the human Cfd1 homologue (NUBP2)
and Cia1 homologue (Ciao1) in cytosolic Fe−S cluster biogenesis remains to be studied.
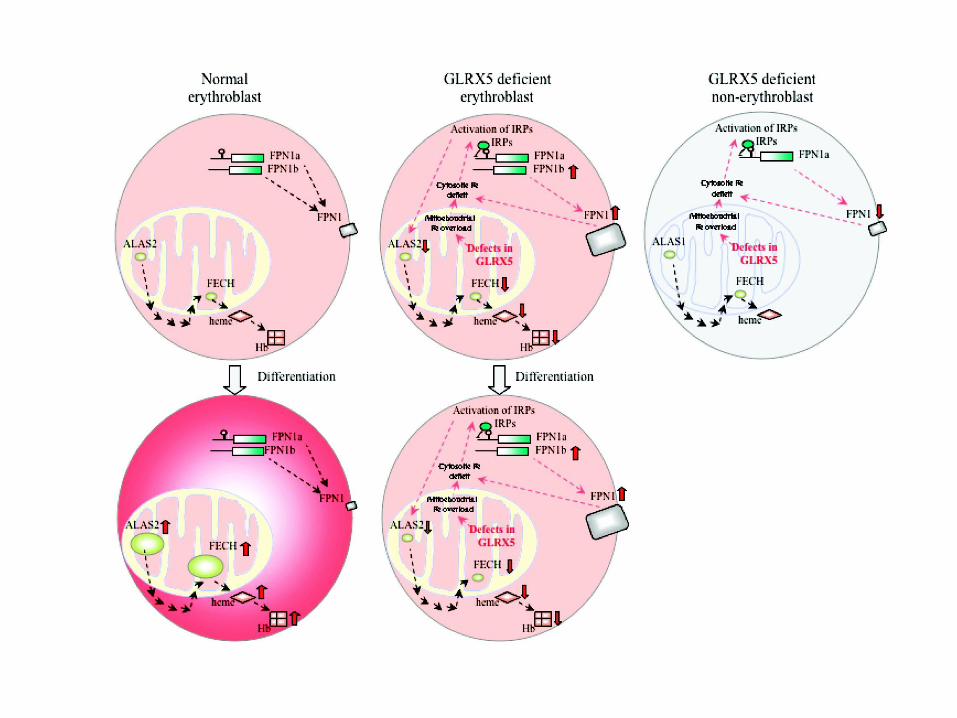

Figure 3. GLRX5 deficiency causes anemia but does not significantly affect non-erythroid tissues. In normal
erythroblasts, ALAS2 and FECH in mitochondria contribute to heme synthesis, which is incorporated into
hemoglobin (Hb). Erythroblasts are the only cells that express ALAS2, which contains a IRE in its 5′ UTR. All
other cells express ALAS1, which is not regulated by the IRE−IRP system. Erythroblasts encode two forms of
the ferroportin (FPN1) transcript, IRE-FPN1a and non-IRE-FPN1b. Both encode an identical FPN1 protein that
functions as the iron exporter. Upon erythroid differentiation, both ALAS2 expression and FECH expression
are upregulated, thus increasing the level of synthesis of heme and hemoglobin, while FPN1 expression is
downregulated (65). In contrast, Fe−S cluster biogenesis is defective in GLRX5-deficient erythroblasts, iron
accumulates in mitochondria, and associated iron deficiency in cytosol activates IRP proteins. The level of
ALAS2 expression is decreased by IRP repression of its 5′ IRE. The level of FECH expression is also
decreased, most likely because FECH does not acquire the Fe−S cluster it needs for stabilization (69). Together,
heme synthesis and subsequent hemoglobinization are inhibited. Levels of expression of both FPN1 transcripts,
including both FPN1a and FPN1b, are increased, perhaps in response to the stress caused by mitochondrial iron
overload. Because FPN1b lacks the IRE, the expression of FPN1b evades the IRP repression and increases the
level of expression of FPN1, which may exacerbate cytosolic iron deficiency. As a result, the GLRX5-deficient
erythroblasts fail to produce enough heme for hemoglobinization upon differentiation. Although GLRX5-
deficient non-erythroblasts also demonstrate iron overload in mitochondria and cytosolic iron deficiency, they
express ALAS1, which does not have IRE and is not repressed by IRPs. Thus, heme synthesis is not
significantly impaired in non-erythroid cell types. In addition, non-erythroblast cells express only FPN1a,
which can be repressed by IRPs as cells develop cytosolic iron depletion.

Figure 4. Example of mitochondrial iron overload and its reversal when iron−sulfur cluster biogenesis is restored.
Shown in the left panels are several fibroblasts that are stained with the Perls’ DAB iron staining technique,
which detects ferric iron and enhances the signal with precipitation of DAB. Iron overload is present in a reticular
pattern consistent with the mitochondrial network of patient fibroblast cells (left), but the iron deposits
disappeared in patient cells rescued by the wild-type GLRX5 gene (right), introduced either by transfection (top)
or by viral transduction (bottom) (32). The scale bars are 10 μm.
