in partial fulfillment of the requirements for
TRANSCRIPT

Synthesis of Guanidinylated-Substituted Polymers that bind Trans-activation Responsive Region of Human Immunodeficiency Virus Type-1 RNA
Master’s Thesis
Presented to
The Faculty of the Graduate School of Arts and Sciences
Brandeis University
Department of Biochemistry
Jason Pontrello, Advisor, Department of Chemistry
Melissa Kosinski-Collins, Advisor, Department of Biology
In Partial Fulfillment
of the Requirements for
Master’s Degree
by
Shakara Lavisha Scott
May, 2013

Copyright by
Shakara L. Scott
© 2013

!!!"
"
This thesis is dedicated to my Godfather James Scott and my best friend Anushka R. Aqil
who always reassured me that the fuel of failure is lack of enthusiasm. But I always
!"#$!#"%&'()'*$+,%-.#'%$'/#'/0')'%/%-1#'2345'/#67
(89/!":';<-.#'="'><%"'*/#?'%"<%'">="!:&'
nor enthusiasm be stirred by spiritless men.
Enthusiasm in our daily work lightens effort and
#+!-:',<=$!'/-#$'@,"<:<-#'#<:A:6BBB7
CJames A. Baldwin

!"#
#
Acknowledgements
There are numerous people without whom this thesis might not have been written and
to whom I am deeply indebted.
To my mentors: It is with immense gratitude that I acknowledge the support and help of
my professors and mentors, Dr. Jason K. Pontrello and Dr. Melissa Kosinski-Collins.
This thesis and fascinating research project would have remained a dream had it not been
for them. The life of an undergraduate research student is not trivial and therefore it was
filled with frustration because nothing works the first time, or the second time. Despite
these difficulties Jason and Melissa attitudes remained steadfast. They both continually
and convincingly conveyed a perennial spirit of adventure and excitement in regard to
research and teaching respectively. In retrospect, I have come to appreciate that they were
preparing me for more than research; but for the ambivalent world out there. They
challenged me to ameliorate my diligence as well as apply myself independently pushing
me towards fulfilling my potential. Thank you, Dr. Pontrello and Dr. KC. I had amazing
learning experience and lots of laughter too!
To my lab buddies: I would be remiss if I did not mention, my lab mentors Nate
Shammay, Larry Friedman, Deb Bordne and Anna Vilenchik. They are all excellent
teachers, scientists and genuinely affable people. Individually, they all played an essential
!"#$%&'%(!)'*+"!,&'-%,$%+!",%)%-)./0%1'2$!-!)21)($%&'("%)%3*",$.4)(5%6",7$($'(%

!"
"
researcher. I admire all of them and I look forward to preserving these life-long
friendships. Thank you, Nate, Larry, Deb and Anna.
To family and friends: Lastly, I want to express my gratitude to my family, and friends
old and new, who have made these four years at Brandeis tolerable and most importantly,
memorable. All of you showered me with your love and support continually. For these
things I am extremely grateful. I would also like to thank Hirvelt Megie for coloring my
life with his candid humor, wealth of hugs and smiles. Thank you Anushka for making
me smile even when you depart from me. I could have never asked for a better best
friend.
!"#$%&'#$(%)&*#+,#-./$0$1&"-2$-(34$#&$#5+,3$%.$)+*4,#6'$7(&-&8(9+-$+,2$&*$
otherwise. Thank you mom for raring a child who is worldly and knowledge driven, I
would not be here without these foundations. Thank you Dad, for always being
optimistic, caring and ever-present! You are the more than a daughter could ever ask for.
I hope you know that I live to make you proud.
Now without further ado, it brings me great joy to present to you my biochemistry
:+'#4*6'$#54'(';$0$9&,'(24*$#5('$1&*3$+$9"-%(,+#(&,$&<$%.$242(9+#(&,$+,2$#5(*'#$<&*$
academics and research.
I hope you enjoy it!
!
!
!
!

ABSTRACT
Synthesis of Guanidinylated-Substituted Polymers that bind Trans-activation Responsive
Region of Human Immunodeficiency Virus Type-1 RNA
A thesis presented to the Department of Biochemistry
Graduate School of Arts and Sciences
Brandeis University
Waltham, Massachusetts
By Shakara Lavisha Scott
Multiple targets exist in the development of HIV-1 anti-viral drugs, one of which
includes the interaction between the transcriptional activator protein (Tat) and the Trans
Activation Response Region (TAR) element of RNA. During transcription, TAR RNA, a
59-base stem-bulge-loop structure, located at the 5’end of all HIV-1 mRNAs, recruits
Tat, which modulates viral gene expression in infected cells. Previous experiments have
shown that the Arginine Rich Motif (ARM) of Tat is integral for the association of Tat to
TAR. Altogether, literature suggests that the inhibition of Tat/TAR RNA interaction is an
attractive route to controlling HIV-1 expression and replication. We sought to design
synthetic polymers that would disrupt the necessary interaction between Tat and TAR-
vi

RNA, hindering HIV replication. To target the TAR-RNA, we sought to replicate the
basic ARM of Tat by functionalizing amine-amenable polymer scaffolds derived from
the Ring-Opening Metathesis Polymerization (ROMP), with the guanidinium derivatives
of arginine and agmatine. To this end, we have successfully synthesized the
guanidinylated polymers. We hypothesize that by amending the polymer scaffolds with
guanidinums, an essential requisite for binding of small molecules to TAR RNA, we may
be able to retard the RNA. To test this hypothesis, we assayed the RNA-binding activities
of the guanidinylated polymers using an Electrophoretic Mobility Shift Assay (EMSA)—
based approach. Our studies indicate that all the synthetic guandiniums are RNA-binding
molecules that recognize and retard the mobility of wild-type TAR RNA in a
concentration range of 1—400 µM. Additionally, we found that the introduction of
magnesium (Mg2+
) in the binding buffer strongly stimulates RNA folding as well as
increases the RNA-binding specificity of the polymeric compounds. To optimize the
binding between the polymers and the RNA we will explore different binding buffers that
may increase the binding affinity, allowing us to characterize the Ka and Kd values. In
addition, it is expected that TAR-RNA binding molecules may inhibit the association of
Tat/TAR; therefore, ongoing work seeks to elucidate the selectivity and specificity of the
guanidinum-conjugated polymers, in addition determining the effects of the polymers on
protein-TAR RNA interactions by EMSA.
vii

!"""#
#
!
!
!
!
!
!
!
!
!
Table of Contents
Dedication iii
Acknowledgements iv-v
Abstract vi-vii
Table of Contents viii-ix
List of Tables x
List of Figures xi-xii
List of Synthetic Schemes xiii
List of Abbreviations xiv-xvi
I. Introduction
Section I: Human Immunodeficiency Virus Type 1 1
HIV-1 Genome 2
The Role of Tat in HIV-1 Replication and Life Cycle 5
Trans-activator of Transcription (TAT) 9
Extracellular Tat 11
Trans-Activator Response (TAR) Element RNA 12
Binding of Tat to Tri-nucleotide Bulge of TAR-RNA 13
Activation of HIV-LTR by Tat 16
Summary 19
Section II: Current Drug Treatments 20
Section III: Motivation
Therapeutic Efforts: Inhibitors Targeting TAR-RNA 22
Small Molecule Inhibitors targeting TAR-RNA 23
Synthetic Polymers Targeting TAR-RNA 26
Summary 29
Section IV: Aims 31
Synthetic Utility of ROMP-derived Polymers 34
Determination of Protein-Nucleic Acid Interactions 34
Summary 35
II. Materials and Methods
Section I: Chemical Synthesis 37
Section II: Biological Assays 49

"$#
#
III. Results
Section I: Chemical Synthesis of Multivalent Guanidiniums 58
Summary 69
Section II: Electrophoretic Mobility Shift Assay (EMSA) 70
IV. Discussion
Section I: Chemical Synthesis of Multivalent Guanidiniums 83
Section II: Electrophoretic Mobility Shift Assay (EMSA) 86
V. Conclusion 94
VI. Future Directions 95
VII. Bibliography 96
VIII. Appendices 102
1H NMR Spectra 102

$#
#
LIST OF TABLES
Page
Table 3.1 Quantified Arginine 10-mer binding experiment 72
Table 3.2 Quantified Arginine 25-mer binding experiment 74
Table 3.3 Quantified Arginine 50-mer binding experiment 76
Table 3.4 Quantified Polymer Binding Experiment 79

$"#
#
LIST OF FIGURES
Page
Figure 1.1 Structure of the HIV-1 Virion 3
Figure 1.2 Organization of HIV-1 Genome and Viral Promoter 5
Figure 1.3 The Essential Steps in Life Cycle HIV-1 7
Figure 1.4 Trans-activator of Transcription (Tat) 10
Figure 1.5 Trans-activation Response Element (TAR) RNA 13
Figure 1.6 Interactions of TAR RNA with the ARM of Tat 14
Figure 1.7 The recognition of HIV-1 TAR RNA by Tat and Cyclin T1 16
Figure 1.8 The activation of RNA polymerase II by Tat and cellular co-factors 18
Figure 1.9 Examples of Antiviral Drugs used to treat HIV 21
Figure 1.10 Classical Approaches to Tat-TAR inhibition 22
Figure 1.11 Structures of small molecules that bind TAR 24
Figure 1.12 Modular design for TAR stem-loop and 3-base-bulge inhibitors 25
Figure 1.13 Neomycin B-Hexaarginine Conjugate 26
Figure 1.14 Structure of TAR RNA binding Oligocarbamate 27
Figure 1.15 Schematic of combination library of branched peptides 28
Figure 1.16 Bindings of FL4 to TAR in the presence of tRNA using EMSA 29
Figure 1.17 A General Scheme for Polymer Conjugation and Design 32
Figure 1.18 Approaches of Various Ligand Displays 35
Figure 3.1 Iodolactonization Mechanism for Isolation of Exo-norbornene 60
Figure 3.2 Mechanism for the formation of the amine-reactive ester 62
Figure 3.3 Gel Electrophoresis of control TAR RNA 71
Figure 3.4 Titration of TAR RNA with ROMP-derived Arginine
Peptidomimetics (10-mer) 73
Figure 3.5 Titration of TAR RNA with ROMP-derived Arginine
Peptidomimetics (25-mer) 75
Figure 3.6 Titration of TAR RNA with ROMP-derived Arginine
Peptidomimetics (50-mer) 77
Figure 3.7 Titration of Arginine-Conjugated polymers with HIV-TAR after pre-
incubation in Mg2+ binding buffer 80
Figure 3.8 Titration of TAR RNA with arginine conjugated 10-mer (A),
norbornene-arginine monomer (B) and free arginine (C) 82
Figure 4.1 Binding-modes of RNA-polymer 88
Figure 4.2 Equilibrium Constant for the binding of ligand to single site 92

$""#
#
LIST OF SCHEMES
Page
Scheme 1 Isolation of Exo-Norbornene 59
Scheme 2 Conversion of Exo-Norbornene to Succimidyl Ester Monomers 61
Scheme 3 Synthesis of Ruthenium Carbene Polymerization Catalyst 63
Scheme 4 Synthesis of Succinimidyl Ester Substituted Polymer Scaffolds 65
Scheme 5 Conjugation of Synthetic Polymer scaffolds with Guanidinium
Derivatives 67
Scheme 6 Synthesis of Arginine Norbornene Control Monomers 68
Scheme 7 General Route for the Synthesis of Agmatine Monomers 69

$"""#
#
LIST OF ABBREVIATIONS
10-mer 10 unit ROMP-derived polymer
25-mer 25 unit ROMP-derived polymer
50-mer 50 unit ROMP-derived Polymer
100-mer 100 unit ROMP-derived polymer
AIDS Autoimmune deficiency syndrome
ARM Arginine Rich Motif
ART Antiretroviral therapy
BIV Bovine Leukemia Virus
CCR5 C-C Chemokine Receptor Type-5
CDCl3 Chloroform
CDK-7 Cyclin-Dependent Kinase-7
CDK-9 Cyclin-Dependent Kinase-9
cDNA Complementary DNA
CH2Cl2 Dichloromethane (a.k.a. methylene
chloride)
C NMR Carbon Nuclear Magnetic Resonance
CTD Carboxyl-Terminal Domain
CXCR4 C-X-C Chemokine receptor Type-4
D2O Deuterium oxide
DEPC Diethyl pyrocarbonate
DMF N, N-dimethylformamide
DMSO Dimethyl sulfoxide
DNA Deoxyribonucleic acid
DSIF DRB Senstivity Inducing Factor
ECM Extracellular Matrix
EDCI N-(3-dimethyl aminopropyl)-!"-ethyl
carbodiimide
EMSA Electrophoretic Mobility Shift Assay
EtO2 Ethyl Ether
Et2O2 Diethyl Ether
EtOAc Ethyl Acetate
FDA Food and Drug Administration
GAGs Glycosaminoglycans
H2O Water

$"!#
#
H2SO4 Sulfuric acid
HAART Highly Active Antiretroviral Therapy
HIV-1 Human Immunodeficency virus type 1 1H NMR Proton Nuclear Magnetic Resonance
HSPGs Heperan Sulfate Proteoglycans
HTLV Human T-Cell Leukemia Virus
I2 Iodine
IN Intergrases
KI Potassium Iodide
KMnO4 Potassium Permanganate
KS Kaposi Sarcoma
LTR Long Terminal Repeat Region
MCH I Major Histocompatibility Class I
MeOH Methanol
MgSO4 Magnesium Sulfate
mRNA Messenger Ribonucliec Acid
N2 Nitrogen
NaHCO3 Sodium Bicarbonate
NaOH Sodium Hydroxide
Na2S2O3 Sodium Thiosulfate
Nef Negative regulator factor
NELF Negative Elongation Factor
NeoR Neomycin B-Hexaarginine
NF-#$%%% Nuclear Factor Kappa B
NHS N-Hydroxysuccinimide
NKT Natural Killer T Cells
NMM N, N-Methylmorpholine
NMR Nuclear magnetic resonance
NRTIs Nucleoside Reverse Transcriptase
Inhibitors
NNRTIs Non-Nucleoside Reverse Transcriptase
Inhibitors
MQ MilliQ
PI Protease Inhibitors
PIC Pre-integration Complex
PPM Parts per million
P-TEFb Positive Transcription Elongation Factor
Complex-b
RNA Ribonucleic Acid
RNA Pol II Ribonucleic Acid Polymerase II

$!#
#
ROMP Ring-opening-metathesis polymerization
SAHA Suberoylanilidehydroxamic Acid
SIV Simian Immunodeficiency Virus
ssRNA Single-stranded Ribonucleic Acid
TAK Tat-associated Kinase
Tat Trans-activator of transcription
TAR Trans-activation Responsive Element
TFIIH Transcription Factor II H
TLC Thin-Layer Chromatography
RT Reverse Transcriptase
VOR Vorinostat

%#
#
I . Introduction:
The synergic interactions of macromolecules in both the extracellular and
intracellular environment are ubiquitous and integral for a variety of pathological and
physiological functions; RNA-protein interactions are a vital class of these protein-nucleic
acid associations. These interactions control some of the most intrinsic biological processes
including activation of cellular genes, transcription, translation, and replication. Owing to the
prevalence of RNA-protein complexations in the cell, regulation of these interactions allows
safeguard and control over the production and proliferation of cellular life. Intensive studies
have shown that RNA-protein mediated interactions play a crucial role in infectious diseases
that are associated with viral replication including cancer, the Human Immunodeficiency
Virus Type-1 (HIV-1), Acquired Immunodeficiency Syndrome (AIDS), and other AIDS
related pathologies such as Kaposi Sarcoma (KS) and Human T-Cell Leukemia Virus
(HTLV) (Dewhurst 1996; Noonan 2000; Mishra 2008; Khalil 2011). As a result, synthetic
methods to selectively control RNA-protein interactions represent attractive ways of
regulating biological functions and can be developed into powerful therapeutic tools.
An archetypal example of protein nucleic acid interactions is the mechanism of trans-
activation in the HIV-&%'()*+,%-%./0'12345%36-3%7-+%8)19(8(3-31:%;4%361%<=>?%8-/:1@(9%0A%
1981 (Zhao 2004; Karn 1999). HIV is a retroviral disease that causes AIDS, a condition that
increases the risk contracting malignant cancers and (/'-+(0/%0A%.0880)3*/(+3(9%(/A193(0/+5%
due to a decimated immune system (Karn 1999; Mishra 2008). In 2012, the global estimate
for people living with HIV/AIDS reached a daunting 34.2 million (UNAIDS 2012). Due to
the lack of a cure, scientists worldwide have been working assiduously to combat this global

&#
#
pandemic with the objective of disrupting the HIV-1 replication cycle (Karn 1998; Mishra
2008).
Early studies of HIV-1 virus revealed significant insight for understanding the viral
replication cycle and the functions of the viral gene products (Cullen 1986; Dingwall et al.
1989; Weeks et. al. 1990; Sodroski 1995a). However, large variability in HIV-1 strains has
complicated development and pursuit of effective antiviral drugs. The most sought out
therapeutic strategy targets the RNA-protein interaction between HIV-1 Transactivator of
transcription (Tat) protein and Transactivation responsive region (TAR) RNA that is
essential to the viral replication and pathogenesis (Karn 1999). On route to a cure is the
design of synthetic drugs that selectively inhibit Tat-TAR interaction, for which a detailed
understanding of the HIV genome and replication cycle is required.
Section I : Human Immunodeficiency V irus Type 1
H I V-1 G enome: The HIV virus belongs to the lentivirus genus and retroviridae
family that also includes the Bovine Leukemia Virus (BIV), Simian Immunodeficiency Virus
(SIV) and HTLV. Retroviruses carry with them a single-stranded RNA and an enzyme that
allows for a reversal of genetic transcription from RNA to DNA. The HIV-1 virus is
spherical in shape and contains two copies of single-stranded RNA (ssRNA) (F igure 1.1).

'#
#
F igure 1.1: Structure of H I V V ir ion. HIV-1 virions possess two strands of genetic material
(viral RNA); viral enzymes encased in a capsid, and are further protected by a protein matrix.
Each virion is spherically shaped and measures about 1/10,000 th of a millimeter in diameter
(Figure not drawn to scale). The enzymes intergrase (IT) and reverse transcriptase (RT) help
the virus copy itself once in the host cell. The outer coat or viral envelope consists of two
layers: a lipid membrane taken from the human cell that the virus particle budded as well as
fatty acids. Protruding through the envelope are HIV proteins Env. The Env protein is made
up of two glycoproteins; the cap, gp120 and the trans-membrane anchor glycoprotein, gp41.
(This image was extracted from NIAID March 29, 2013
http://www.niaid.nih.gov/topics/hivaids/understanding/biology/Pages/structure.aspx).
Each copy of RNA is approximately 10, 000 nucleotides in length and encodes for nine genes
(gag, pol, vif, vpr, rev, tat, vpu, env and nef) (F igure 1.2). Of the nine genes, six encode for
viral accessory proteins, which assist in the proliferation of the HIV-1 virus. The HIV viral
proteins vif and vpu influence the assembly and budding of new virions. Env encodes for the
viral envelope glycoprotein SU (gp160) that is essential for the binding of host cell receptors
and co-receptors. Nef, the negative regulator factor protein participates in cell activation, T-
cell apoptosis and the down-regulation of host molecules that are critical for the development
of cellular and humoral immune responses (Ranjan Das et al. 2005). Rev mediates the
transportation of unspliced messenger RNA (mRNA) from the nucleus into the cytoplasm.

(#
#
The molecular functions of viral protein R (vpr) include nuclear import of viral pre-
integration complex (PIC), modulation of T-cell apoptosis, transcriptional co-activation of
viral and host genes, and regulation of nuclear factor kappa B (NF-#$B%-93('(34%CD0E-/%
2011). Tat is a Trans-activating protein that regulates viral replication and gene expression.
Taken together, though all the viral proteins contribute to the processes that fuel the HIV-1
infection and evasion of the immune system, the role of Tat, Rev, and Vpr are considered to
be the largest contributors to the morbidity and mortality of HIV/AIDS (Karn 1999; Romani
2010; Kogan 2011). The numerous functions of Vpr, Rev and Tat in the viral life cycle
suggest that they would be attractive targets for therapeutic intervention and development of
HIV antiviral agents. In this manuscript, we focus on the Tat protein.
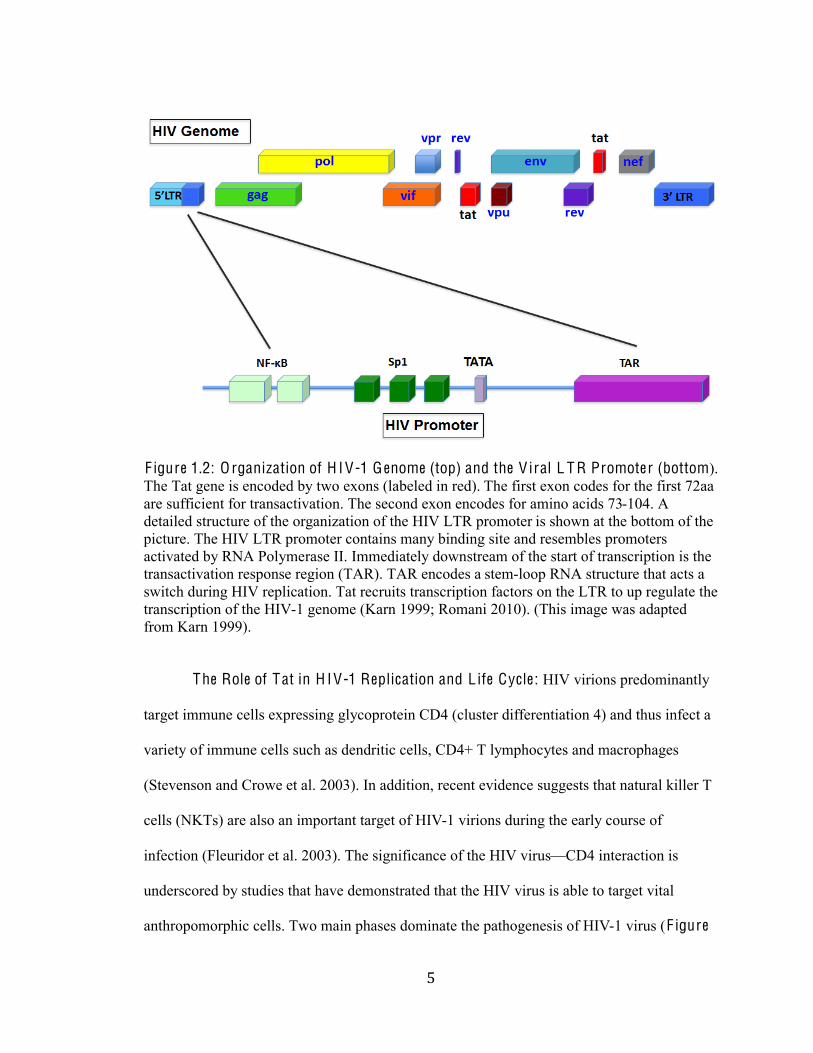
)#
#
F igure 1.2: O rganization of H I V-1 G enome (top) and the V iral L T R Promoter (bottom).
The Tat gene is encoded by two exons (labeled in red). The first exon codes for the first 72aa
are sufficient for transactivation. The second exon encodes for amino acids 73-104. A
detailed structure of the organization of the HIV LTR promoter is shown at the bottom of the
picture. The HIV LTR promoter contains many binding site and resembles promoters
activated by RNA Polymerase II. Immediately downstream of the start of transcription is the
transactivation response region (TAR). TAR encodes a stem-loop RNA structure that acts a
switch during HIV replication. Tat recruits transcription factors on the LTR to up regulate the
transcription of the HIV-1 genome (Karn 1999; Romani 2010). (This image was adapted
from Karn 1999).
The Role of Tat in H IV-1 Replication and L ife Cycle : HIV virions predominantly
target immune cells expressing glycoprotein CD4 (cluster differentiation 4) and thus infect a
variety of immune cells such as dendritic cells, CD4+ T lymphocytes and macrophages
(Stevenson and Crowe et al. 2003). In addition, recent evidence suggests that natural killer T
cells (NKTs) are also an important target of HIV-1 virions during the early course of
infection (Fleuridor et al. 2003). The significance of the HIV virusFCD4 interaction is
underscored by studies that have demonstrated that the HIV virus is able to target vital
anthropomorphic cells. Two main phases dominate the pathogenesis of HIV-1 virus (F igure

*#
#
1.3) (Karn 1999). In the first phase, the virus enters the cell via a fusion mechanism between
the glycoprotein 120 SU (gp120) envelope of the virion and the CD4 cell membrane receptor.
This fusion between the virus and the host cell membrane also requires chemokine
coreceptors CCR5 (predominant during acute and asymptomatic phases of the HIV-1
infection) and CXCR4 (Crowe 2003; Stevenson et al. 2003; Mishra 2008). Once in the
cytosol, the virus uncoats and uses its inherent reverse transcriptase (RT) to synthesize
double-stranded viral DNA. This is followed by nuclear import of the viral DNA. The
accessory protein Rev transports the viral DNA into the nucleus where intergrase (IN)
catalyzes the integration to the host genome (Mishra 2008). The second phase involves viral
gene expression, replication, assembly, and virion maturation (Karn 1999).
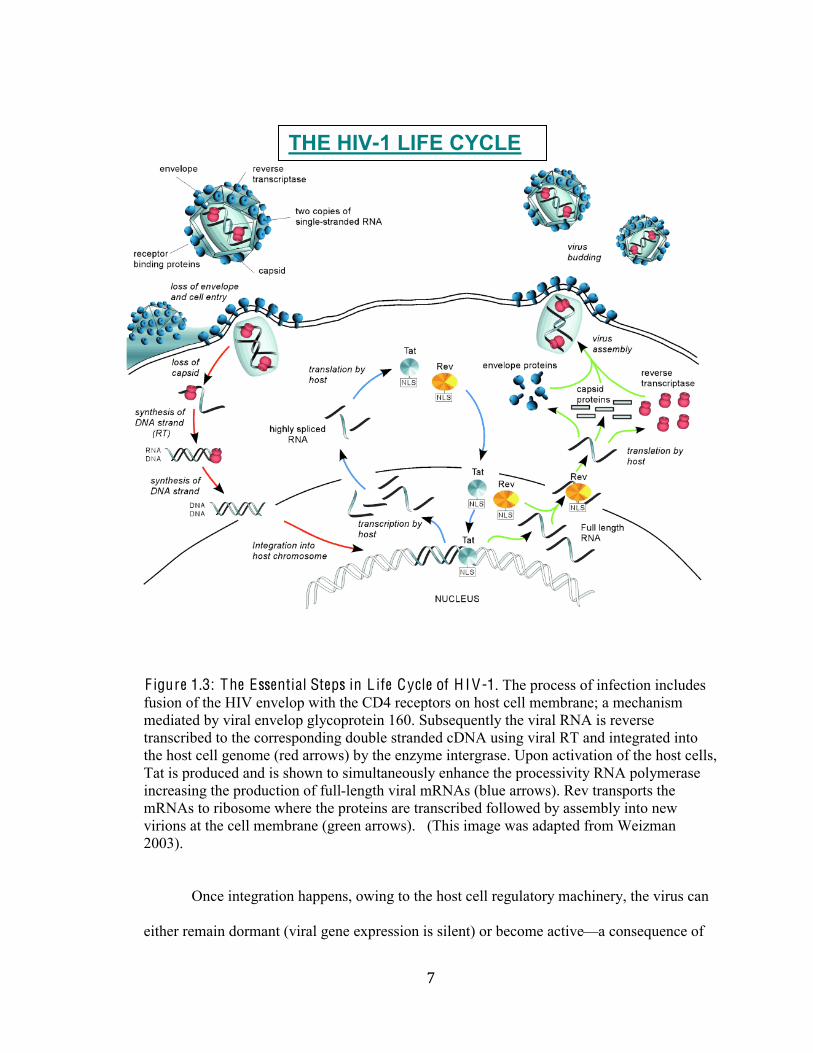
+#
#
F igure 1.3: The Essential Steps in L ife Cycle of H I V-1. The process of infection includes
fusion of the HIV envelop with the CD4 receptors on host cell membrane; a mechanism
mediated by viral envelop glycoprotein 160. Subsequently the viral RNA is reverse
transcribed to the corresponding double stranded cDNA using viral RT and integrated into
the host cell genome (red arrows) by the enzyme intergrase. Upon activation of the host cells,
Tat is produced and is shown to simultaneously enhance the processivity RNA polymerase
increasing the production of full-length viral mRNAs (blue arrows). Rev transports the
mRNAs to ribosome where the proteins are transcribed followed by assembly into new
virions at the cell membrane (green arrows). (This image was adapted from Weizman
2003).
Once integration happens, owing to the host cell regulatory machinery, the virus can
either remain dormant (viral gene expression is silent) or become activeFa consequence of
!"#$"%&-'$(%)#$*+*(#

,#
#
stimulating infected host cells with mitogens (Karn 1999). The activation of transcription for
the proviral genome is regulated by transcription factors: NF-#$ and Sp1 and the Tat protein
(F igure 1.3). The HIV pre-mRNA that is transcribed from the proviral DNA contains several
splicing signals (Mishra 2008). In the nascent stages of the HIV replication cycle mostly 2 kb
mRNA transcripts to be produced (F igure 1.3). These mRNA transcripts are translated into
regulatory proteins: Tat, Nef and Rev (Mishra 2008; Romani 2010). The Tat protein is
imported in the nucleus where it binds to nascent RNA transcript (TAR RNA) and with the
help of Tat-associated kinases (TAK), dramatically stimulates transcription elongation and
increases the production of mRNA transcripts (Karn 1999; Stevenson 2003; Weizman 2003;
Mishra 2008). In order for the lifecycle to shift to the late phases, the production of unspliced
pre-mRNA transcripts are needed for assembly into the progeny virions. Moreover, in order
for HIV to produce its complete range of structural, accessory enzymatic proteins, unspliced
~9 kb and singly spliced ~4 kb transcripts are required (Mishra 2008). Once these unspliced
and singly spliced transcripts are generated they are translocated to the cytoplasm and
ribosomes by viral protein Rev with the help of host cell nuclear export machinery (F igure
1.3).
At the ribosomes, the unspliced RNA transcripts are translated into Gag and Gal-Pol
proteins, while the unspliced RNA is translated into Env, Vpu, Vif, and Vpr. Finally, new
progeny virions are packaged and released through the cell membrane surface of the host cell
by budding (F igure 1.3). Viral proteins Nef and Env mediate the budding mechanism;
degrading and down regulating cell surface CD4, thus avoiding immune response. This
stealthy release of new progeny into the interstitial of the body allows the virus to be
metastasized to other cells without detection and perpetuates the progression to AIDS and
decimated immune systems.

-#
#
T rans-activator of T ranscr iption: Tat is one of the six HIV-1 regulatory protein
products essential for transactivation of viral and cellular genes. It is expressed in both the
early and late stages of the viral replication cycle. Tat that is released in the nascent stages of
replication is found in both the nuclei and nucleolus of HIV infected cells; when it is
produced in the later stages, Tat is predominantly found in the extracellular environment. Tat
has a variable length of 86-104 amino acids and is encoded by two exons Fdepending on the
viral strain. The first-exon form encodes the first 72 amino acids, which are sufficient for Tat
transactivation. The second exon codes for amino acids 73-104. Moreover, the two-exon
form has an additional carboxyl terminal that, based upon the viral isolate varies in length
between 86 and 104 amino acids; the additional amino acids are appended at the carboxyl
terminal (Weissman et al. 1998; Jeang 1996; Aboul-ela et al. 1999). The generation of these
two forms of Tat is regulated during translation via splicing mechanisms: the 86 amino acid
version is produced from completely spliced mRNA and the 104 amino acid version from
partially spliced HIV mRNA transcripts (Weissman et al. 1999; Amendt et al. and Bilodeau
et al. 1999). Consequently, the one-exon form of Tat is expressed predominantly during the
nascent stages while the two-exon version of Tat materializes in the later stages (Amendt et
al. 1994; Romani 2010).
There are five structural regions of the Tat protein: the N-terminal domain, which
contains amino acids 1-20, the cysteine rich region that contains seven high conserved
cysteine residues (residues 22-37), the core region (amino acids 37-48), the basic region
(residues 48-72) and the carboxyl terminal domain (C-terminal; residues 72-86) (F igure 1.4).

%.#
#
F igure 1.4: T rans-activator of T ranscr iption (Tat). L eft: Organization of Tat peptide. Right: Primary structure of HIV-1 Tat peptide with the Arginine-Rich Motif (ARM) residues
48-57 in red (Yang 2005). L eft: Primary structure of HIV-1 Tat peptide.
(The image on right was adapted and modified from Yang 2005).
Previous work has indicated that deletion and substitution experiments of residues in
the Cys-rich region have resulted in loss of trans-activation; suggesting that it is required for
Tat function (formation of intra-molecular disulphide bonds) but they are not directly
involved in TAR recognition (Aboul-ela 1999; Yang 2005). The basic region, which consists

%%#
#
of Arginine Rich Motif (ARM), is conserved over several strains of the HIV-1 and regulates
the Transactivation activity of Tat. Furthermore, the basic region is an essential requisite for
the interactions between the protein and its nucleic acid conjugate, TAR RNA (Yang 2005).
In addition, some have discovered that the carboxyl-terminal domain (CTD) of Tat represses
the transcription of major histocompatibility class I genes (MCH I), which are the first line of
cell immune defense (Weissman 1998). Overall, Tat is a multifunctional protein that has
significant effects on both the virus and the host cell genes.
Extracellular Tat: In addition to intracellular Tat that activates HIV LTR, Tat is also
found in the extracellular matrix. Extracellular Tat along with helper gp120, are viral
products secreted by HIV-1 infected T-cells in the extracellular environment (Bugatti 2007;
Romani 2010). Cohesively, they act as immune-suppressors, activating quiescent T-cells and
targeting HIV-nonpermissive cells/non-HIV-infected cells for progression of the HIV-1
infection (Litovchick 2001; Bugatti 2007). A compilation of research studies elucidates the
entrance of extracellular Tat into cells via an endocytic pathway by binding to an invariable
amount of cell surface receptors, including vascular endothelial growth factor, heparan
sulfate proteoglycan chemokine receptors CCR2, CCR3 and CXCR4 (Xiao et al. 2000;
Bugatti 2007), and heparan sulfate proteoglycans (HSPGs) (Tyagi et al. 2001; Bugatti 2007).
The bindings of Tat by these receptors increase its local concentration in the extracellular
matrix (ECM) and mediate its internalization and trans-activating activity (Noonan 1996;
Vendeville 2004; Bugatti 2007; Miyauchi 2009). Studies of Tat-derived peptides have
demonstrated that residues 48-60 from the basic domain (protein transduction domain or
PTD) accounts for the functional internalization into cells (Buggati 2007; Romani 2010).
Furthermore, Tat contributes to the development of AIDS and other AIDS-associated
pathologies by concomitantly inducing oxidative stress in the blood-brain barrier cells (ECs)
(Price 2005) and causing apoptosis in cardiomyocytes (Fiala 2004), neurons (Singh 2004)

%&#
#
and other immune cells. For example, Tat enters host macrophages and inhibits nitric oxide
synthase gene activity. This inhibitory effect of Tat on the production of nitric oxide renders
the host vulnerable to infections, since nitric oxide provides the first line of defense against
opportunistic pathogens (Romani 2010).
T rans-activation Response E lement (T A R) RN A : Replication of HIV-1 LTR
requires Tat to bind to trans-activation response element (TAR) RNA, a conserved 59-base
stem-2008%+3)*93*)1%209-31:%(/%361%20/E%31)@(/-2%)1E(0/%CGHIB%-3%361%J"%1/:%0A%-22%K=L-1
mRNA transcripts F igure 1.2 (Yang 2005). Several studies performed using mutant HIV-1
variants indicate that the Tat protein and the TAR RNA sequence are necessary for viral
replication and pathogenesis (Jeang et al. 1999; Karn 1999; Harrich et al. 1995). The
structural components of TAR RNA, spanning from nucleotides +1 to +57 (F igure 1.5)
includes: the stem-loop, upper arm, 3-base bulge, and the lower stem (Karn 1999; Yang
2005). The 3- base-bulge along with two base pairs above and below the bulge constitutes the
core elements for Tat binding (Yang 2005). Research has shown that the U-rich 3 base-bulge
residues (U 23, C24, U25 or UUU) near the apex of the TAR RNA stem are necessary for
specific binding and recognition of the Tat protein in vivo trans-activation. The mutations in
TAR RNA that affect the structure and base pairing in the U-rich bulge completely abolish
Tat association.
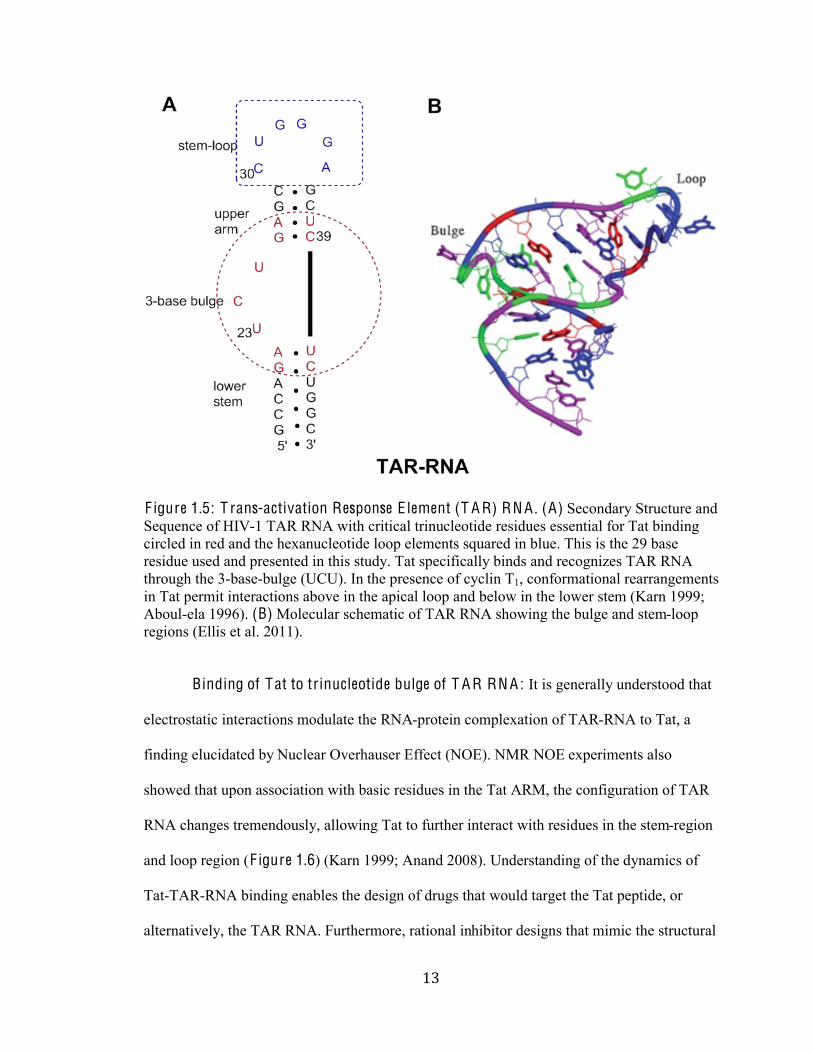
%'#
#
F igure 1.5: T rans-activation Response E lement (T A R) RN A . (A) Secondary Structure and
Sequence of HIV-1 TAR RNA with critical trinucleotide residues essential for Tat binding
circled in red and the hexanucleotide loop elements squared in blue. This is the 29 base
residue used and presented in this study. Tat specifically binds and recognizes TAR RNA
through the 3-base-bulge (UCU). In the presence of cyclin T1, conformational rearrangements
in Tat permit interactions above in the apical loop and below in the lower stem (Karn 1999;
Aboul-ela 1996). (B) Molecular schematic of TAR RNA showing the bulge and stem-loop
regions (Ellis et al. 2011).
Binding of Tat to trinucleotide bulge of T A R RN A : It is generally understood that
electrostatic interactions modulate the RNA-protein complexation of TAR-RNA to Tat, a
finding elucidated by!Nuclear Overhauser Effect (NOE). NMR NOE experiments also
showed that upon association with basic residues in the Tat ARM, the configuration of TAR
RNA changes tremendously, allowing Tat to further interact with residues in the stem-region
and loop region (F igure 1.6) (Karn 1999; Anand 2008). Understanding of the dynamics of
Tat-TAR-RNA binding enables the design of drugs that would target the Tat peptide, or
alternatively, the TAR RNA. Furthermore, rational inhibitor designs that mimic the structural
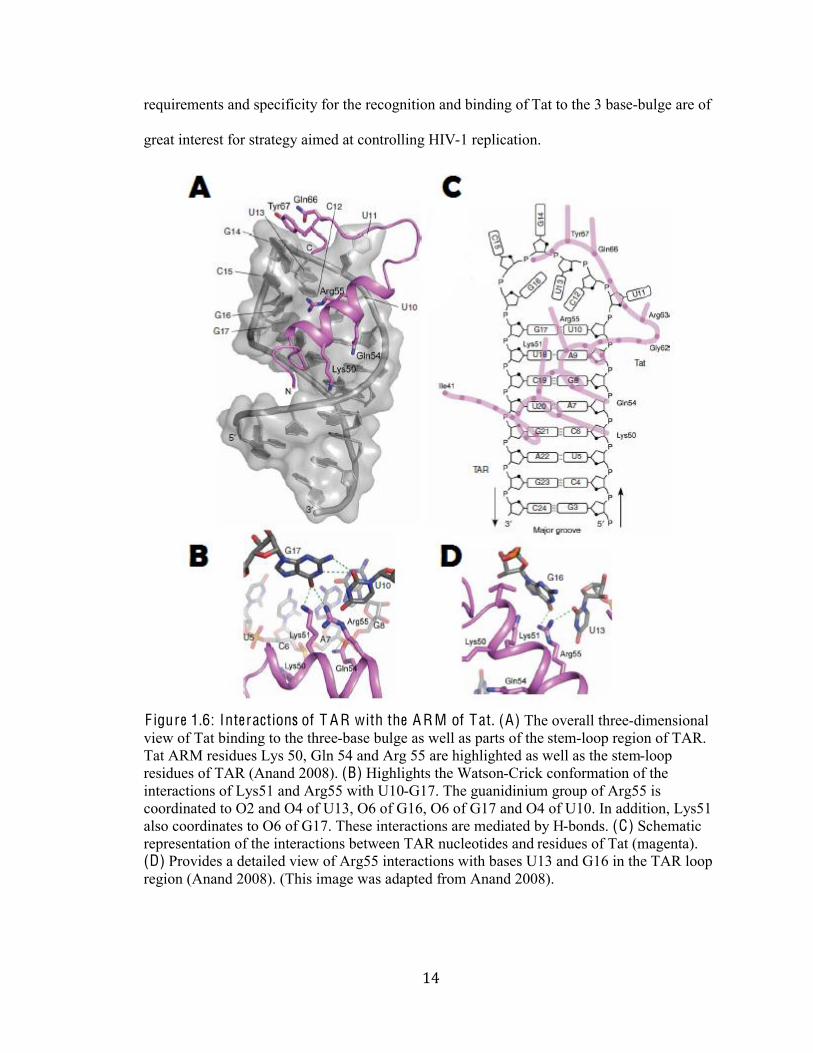
%(#
#
requirements and specificity for the recognition and binding of Tat to the 3 base-bulge are of
great interest for strategy aimed at controlling HIV-1 replication.
F igure 1.6: Interactions of T A R with the A R M of Tat. (A) The overall three-dimensional
view of Tat binding to the three-base bulge as well as parts of the stem-loop region of TAR.
Tat ARM residues Lys 50, Gln 54 and Arg 55 are highlighted as well as the stem-loop
residues of TAR (Anand 2008). (B) Highlights the Watson-Crick conformation of the
interactions of Lys51 and Arg55 with U10-G17. The guanidinium group of Arg55 is
coordinated to O2 and O4 of U13, O6 of G16, O6 of G17 and O4 of U10. In addition, Lys51
also coordinates to O6 of G17. These interactions are mediated by H-bonds. (C) Schematic
representation of the interactions between TAR nucleotides and residues of Tat (magenta).
(D) Provides a detailed view of Arg55 interactions with bases U13 and G16 in the TAR loop
region (Anand 2008). (This image was adapted from Anand 2008).

%)#
#
Mutational studies have identified that in addition to acting as the binding site for
Tat, the TAR acts as the recognition signal for Tat cellular cofactor cyclin T1 (CycT1) a
component of the Tat-associated kinase (TAK)/Positive Elongation Factor (P-TEFb) CTD
kinase complex (Garber 1998b; Karn 1999; Raghunathan 2006). The CylcT1 once recruited
by Tat binds the apical stem loop sequence of TAR. It is important to note that, the binding of
the stem-loop sequence by the cofactor cyclin T1 (F igure 1.7) is required only for trans-
activation, but not for Tat binding (Karn 1999). Therefore, interfering with the interaction
between the Tat/CycT1 complexes can also be an attractive target for developing HIV-1
antiviral agents.
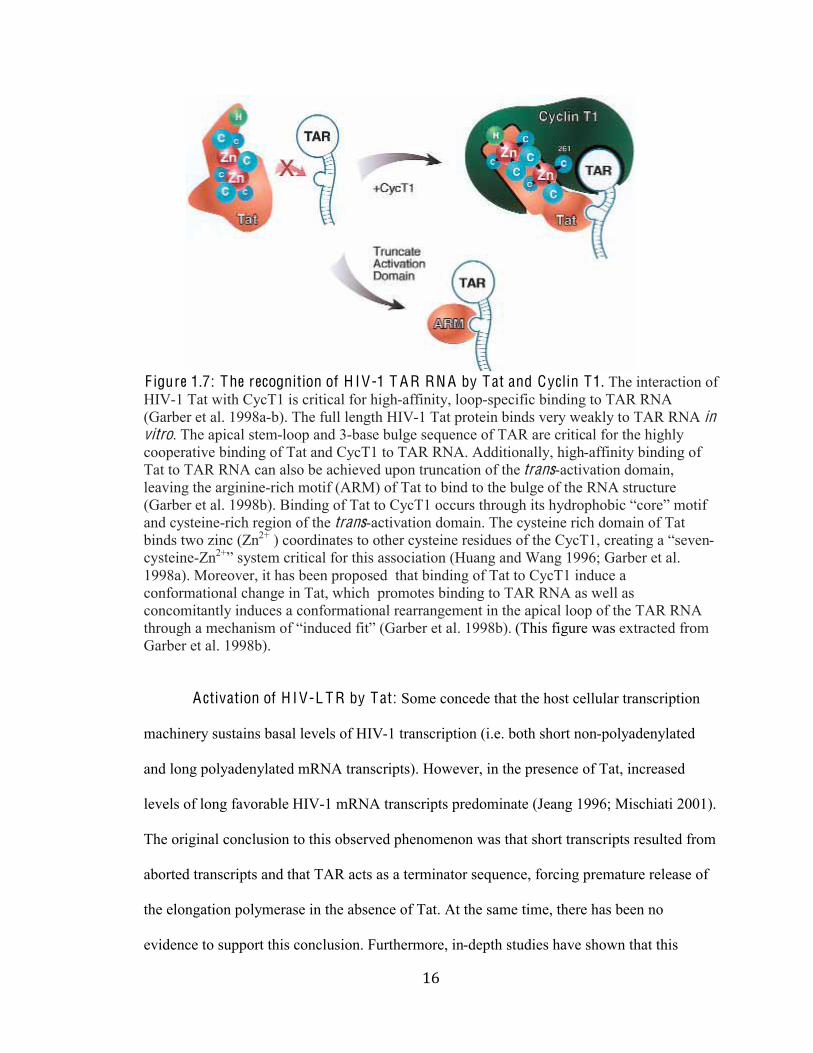
%*#
#
F igure 1.7: The recognition of H IV-1 T A R RN A by Tat and Cyclin T1. The interaction of
HIV-1 Tat with CycT1 is critical for high-affinity, loop-specific binding to TAR RNA
(Garber et al. 1998a-b). The full length HIV-1 Tat protein binds very weakly to TAR RNA in vitro. The apical stem-loop and 3-base bulge sequence of TAR are critical for the highly
cooperative binding of Tat and CycT1 to TAR RNA. Additionally, high-affinity binding of
Tat to TAR RNA can also be achieved upon truncation of the trans-activation domain,
leaving the arginine-rich motif (ARM) of Tat to bind to the bulge of the RNA structure
CM-);1)%13%-2N%&OOP;BN%$(/:(/E%0A%H-3%30%Q49H&%099*)+%36)0*E6%(3+%64:)0860;(9%.90)15%@03(A%
and cysteine-rich region of the trans-activation domain. The cysteine rich domain of Tat
binds two zinc (Zn2+ ) coordinates to other cysteine re+(:*1+%0A%361%Q49H&,%9)1-3(/E%-%.+1'1/-
cysteine-Zn2+5%+4+31@%9)(3(9-2%A0)%36(+%-++09(-3(0/%CK*-/E%-/:%R-/E%&OOST%M-);1)%13%-2N%
1998a). Moreover, it has been proposed that binding of Tat to CycT1 induce a
conformational change in Tat, which promotes binding to TAR RNA as well as
concomitantly induces a conformational rearrangement in the apical loop of the TAR RNA
36)0*E6%-%@196-/(+@%0A%.(/:*91:%A(35%CM-);1)%13%-2N%&OOP;BN%(This figure was extracted from
Garber et al. 1998b).
Activation of H I V-L T R by Tat: Some concede that the host cellular transcription
machinery sustains basal levels of HIV-1 transcription (i.e. both short non-polyadenylated
and long polyadenylated mRNA transcripts). However, in the presence of Tat, increased
levels of long favorable HIV-1 mRNA transcripts predominate (Jeang 1996; Mischiati 2001).
The original conclusion to this observed phenomenon was that short transcripts resulted from
aborted transcripts and that TAR acts as a terminator sequence, forcing premature release of
the elongation polymerase in the absence of Tat. At the same time, there has been no
evidence to support this conclusion. Furthermore, in-depth studies have shown that this

%+#
#
phenomenon occurs because TAR acts as a pause site that result in a brief kinetic block to
transcription (Muesing et al. 1987; Selby et al. 1989; Karn 1999). In the presence of Tat the
kinetic block is deactivated and transcription of viral LTR occurs.
In HIV-1 infected cells, the first step in activation of the HIV-1 LTR is the
recruitment of RNA polymerase II (RNA Pol II) (F igure 1.8). Once the RNA Pol II, along
with its mediators that regulate the carboxyl-terminal domain (CTD) of the enzyme is bound,
several downstream events must occur. The phosphorylation of the CTD by the Cylcin-
Dependent Kinase-7 (CDK-7) component of the Transcription factor II H (TFIIH) complex
allows the RNA POL II to clear the promoter and begin the transcription of TAR. Soon after
initiation and transcription of TAR, RNA Pol II is stalled by the repressive Negative
Elongation Factor (NELF), another component of basal transcription factor TFIIH. The
nascent RNA chain folds into the TAR RNA structure constituted of the 3-base bulge and
apical stem-loop. In order to reinitiate transcription, the HIV regulatory protein Tat is
recruited to the three-base bulge sequence of TAR and subsequently recruits the positive
transcription elongation factor complex b (P-TEFb)/Tat-associated kinase (TAK). The P-
TEFb complex consists of CDK9 and Cyclin T1. Tat interacts directly with the cyclin T1
subunit of P-TEFb through zinc (Zn2+) cation to induce the cooperative binding of Cylcin-
Dependent Kinase-9 (CDK-9) (F igure 1.8). This recruitment enables the phosphorylation of
the negative elongation factors as well as the CTD of RNA Pol II, which allows the RNA Pol
II to transcribe the remainder of the HIV-1 genome (Karn 1999). Furthermore, Tat binding
enhances the processivity of the RNA Polymerase II (RNA Pol II) elongation complex,
which induces transcription of HIV-1 long terminal region (LTR) (Karn 1999).
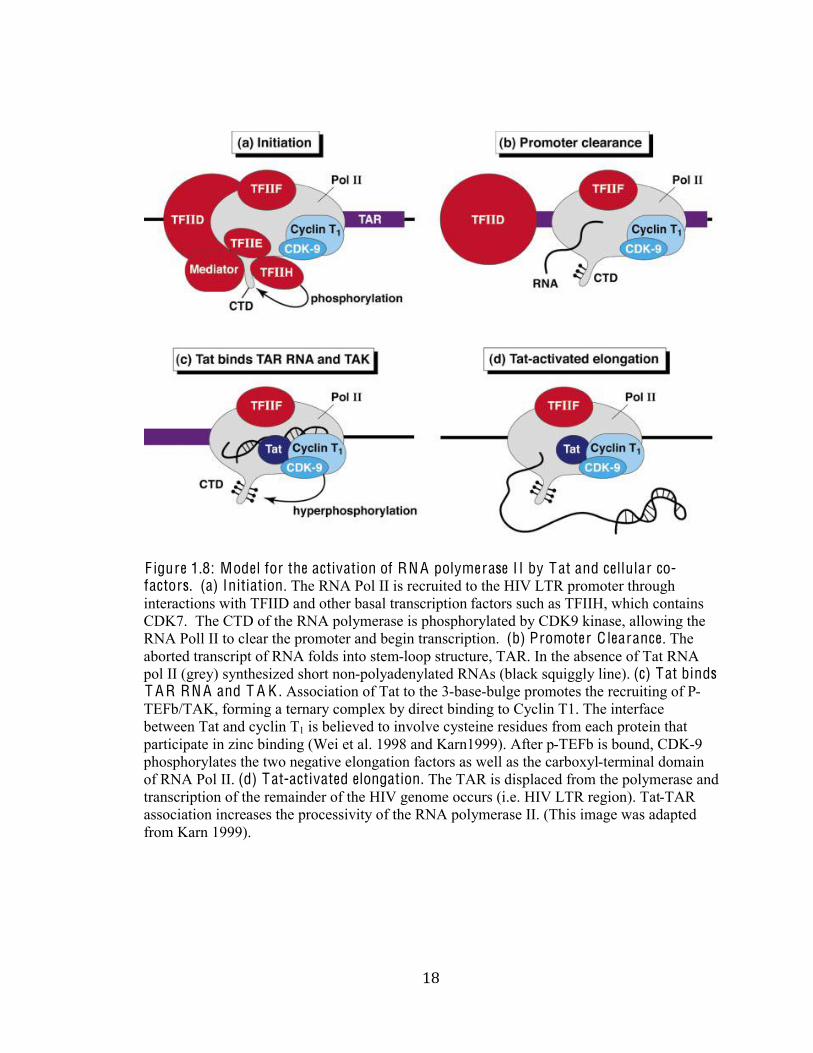
%,#
#
F igure 1.8: Model for the activation of RN A polymerase I I by Tat and cellular co-factors. (a) Initiation. The RNA Pol II is recruited to the HIV LTR promoter through
interactions with TFIID and other basal transcription factors such as TFIIH, which contains
CDK7. The CTD of the RNA polymerase is phosphorylated by CDK9 kinase, allowing the
RNA Poll II to clear the promoter and begin transcription. (b) Promoter C learance. The
aborted transcript of RNA folds into stem-loop structure, TAR. In the absence of Tat RNA
pol II (grey) synthesized short non-polyadenylated RNAs (black squiggly line). (c) Tat binds T A R RN A and T A K . Association of Tat to the 3-base-bulge promotes the recruiting of P-
TEFb/TAK, forming a ternary complex by direct binding to Cyclin T1. The interface
between Tat and cyclin T1 is believed to involve cysteine residues from each protein that
participate in zinc binding (Wei et al. 1998 and Karn1999). After p-TEFb is bound, CDK-9
phosphorylates the two negative elongation factors as well as the carboxyl-terminal domain
of RNA Pol II. (d) Tat-activated elongation. The TAR is displaced from the polymerase and
transcription of the remainder of the HIV genome occurs (i.e. HIV LTR region). Tat-TAR
association increases the processivity of the RNA polymerase II. (This image was adapted
from Karn 1999).

%-#
#
Summary: Altogether, the data available in the literature suggest that inhibition of
Tat/TAR RNA interactions and CyclinT1/TAR interaction could be of great interest for
controlling HIV-1 replication. Accordingly, this knowledge has catalyzed the search for
molecular compounds that specifically block Tat/TAR interactions. In this study we focus on
elucidating the binding of synthetic to the TAR-RNA to further develop a TAR-RNA drug
that may warrant pharmaceutical development.

&.#
#
Section I I . Cur rent Drug T reatments:
There is currently no cure for HIV. Yet, the HIV pandemic remains one of the most
deadly threats to world health and presents a significant development challenge (Karn 1999;
UNAIDS 2012). There are approximately thirty-three million people living with HIV/AIDS
worldwide. However, in the thirty-two years since the discovery of HIV, only twenty-five
antiviral drugs are available, for mass use and production. These drugs have been able to
reduce HIV prevalence rates but they are by no means effective preventions or cures for the
disease.
Typically the regulation of HIV includes antiretroviral therapy (ART) and Highly
Active Antiretroviral Therapy (HAART). There are over twenty-three U.S. Food and Drug
Administration (FDA) approved antiretroviral drugs that are used to treat the disease. The
function of ARTs is to repress the growth and reproduction of HIV as well as allow people
infected to live longer, healthier lives. Using several of these drugs in combination also
allows for the rebuilding of the immune system. These drugs are classified by the phase of
the retrovirus life cycle that the drug inhibits; the seven categories are as follows: Entry
inhibitors, CCR5 receptors antagonist, Nucleoside Reverse Transcriptase Inhibitors (NRTIs),
Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs), Protease Inhibitors (PIs), Fusion
Inhibitors (FIs), and Intergrase Inhibitors (IIs). However, these drugs have a variety of
adverse side effects, which makes selecting a regiment complex and variable among
individuals. In addition, although HIV chemotherapy inhibits most viral replication, there is
still a remaining population of latently infected cells that remain unaffected.
Viral latency is one of the aspects of the virus that makes it difficult to cure. HIV
viral latency is the ability of the virus to integrate into resting T-Cells and other cellular
reservoirs (Stevenson, 2003). In the case that the cells are activated, viral production spirals

&%#
#
off. A currently studied therapeutic approach involves activating resting cells and flushing
the virus out of hiding, making it vulnerable to antiretroviral drugs and the natural immune
response. The most recent drug that induces the expression of HIV RNA and genomes in
resting CD4+ cells is a histone deacetylase inhibitor, suberoylanilidehydroxamic acid SAHA
also known as vorinostat, VOR (F igure 1.9a). Histone deacetylases are recruited to the HIV
long terminal repeat (LTR) promoter, and are therefore one of the several restrictions that can
limit LTR expression and maintain viral latency (Archin et al. 2012).
F igure 1.9: Examples of Antiviral Drugs used to treat H I V . (A) Vorinostat is a histone
deacetylase inhibitor that acts on HIV-1 infected CD4+ cells inducing HIV RNA and genome
expression. (B) Maraviroc is an entry inhibitor as well as a chemokine receptor CCR5
antagonist, preventing HIV gp 160 proteins from associating with the cell. The chemokine
receptor CCR5 is an essential co-receptor is an essential co-receptor for a majority of HIV
strains and is necessary for the entry process of the virus into the host c

&&#
#
Motivation and A ims:
Therapeutic E fforts: Inhibitors Targeting T A R-RN A: There are two bimolecular
strategies to inhibiting the Tat/TAR-RNA complex: antibiotic and small molecule analogues
that are able to selectively bind to the TAR-RNA and those that interact with the Tat ARM
(F igure 1.10). Pharmological compounds that have already been developed to bind to TAR-
RNA vary in structural components and fall into three subcategories: those that bind to the
UCU or UUU trinucleotide bulge tightly and consequently outcompete the endogenous
protein partner, Tat peptide; those that bind the 3-base-bulge together with either lower or
upper stem-loop region; and those that bind the stem-loop structure preventing trans-
activation, by impeding the TAR-RNA interaction with Cyclin T1 (Yang 2005).
F igure 1.10: C lassical Approaches to Tat-T A R inhibition. Class I inhibitors that bind
TAR-RNA and Class II that bind the Tat protein outcompeting endogenous is cognate RNA
partner. The blue circle and green Pac-Man structure represents the small molecules designed
to target Tat and TAR-RNA respectively.
Tat
TAR-RNA TAR-RNA
Tat
Tat
TAR-RNA
Tat
TAR-RNA
TAR-RNA
Tat
Tat
TAR-RNA
Uninhibited
Class I
Class II

&'#
#
Small molecule Inhibitors targeting T A R RN A : =/%361%2-31%&OOU"+,%-/%(/'-)(-;24%
-@0*/3%0A%/0/8183(:(9%+@-22%@0219*21+%CVR%W%JUUU>-B%71)1%:(+90'1)1:%30%(/6(;(3%H-3XH<I%
complex formation. Molecules as small as argininamide, were of the first shown to inhibit the
association of TAR RNA to Tat protein. In fact it is considered one of the best-investigated
TAR RNA ligands (Krebs 2003; Thomas 2008). It also binds at the bulge region by via H-
bonding to the UCU residues as well as residue G26 in the stem (F igure 1.11a). These early
findings paved the way for the development of other molecular inhibitors of TAR that
possessed guanidine moieties. To enhance the affinity, or direct the specificity, argininamide
bifunctional ligands consisting of ethidium bromide (known intercalator of DNA) and
arginine were developed. An example of these arginine conjugates is shown in F igure 1.11b.
This ethidium-arginine conjugate inhibits the association of Tat/TAR by binding the
trinucleotide bulge of TAR RNA with high affinity. The basic guanidinium groups of both
arginine analogues are said to interact with the phosphate ion backbone of TAR RNA, while
361%90/Y*E-31:%Z-rings have been hypothesized to participate in ! " ! interactions. This
incorporation of the aromatic rings and therefore ! " ! interactions increases the strength
and specificity of the guanidinum compoundFserving as a pseudo anchor.
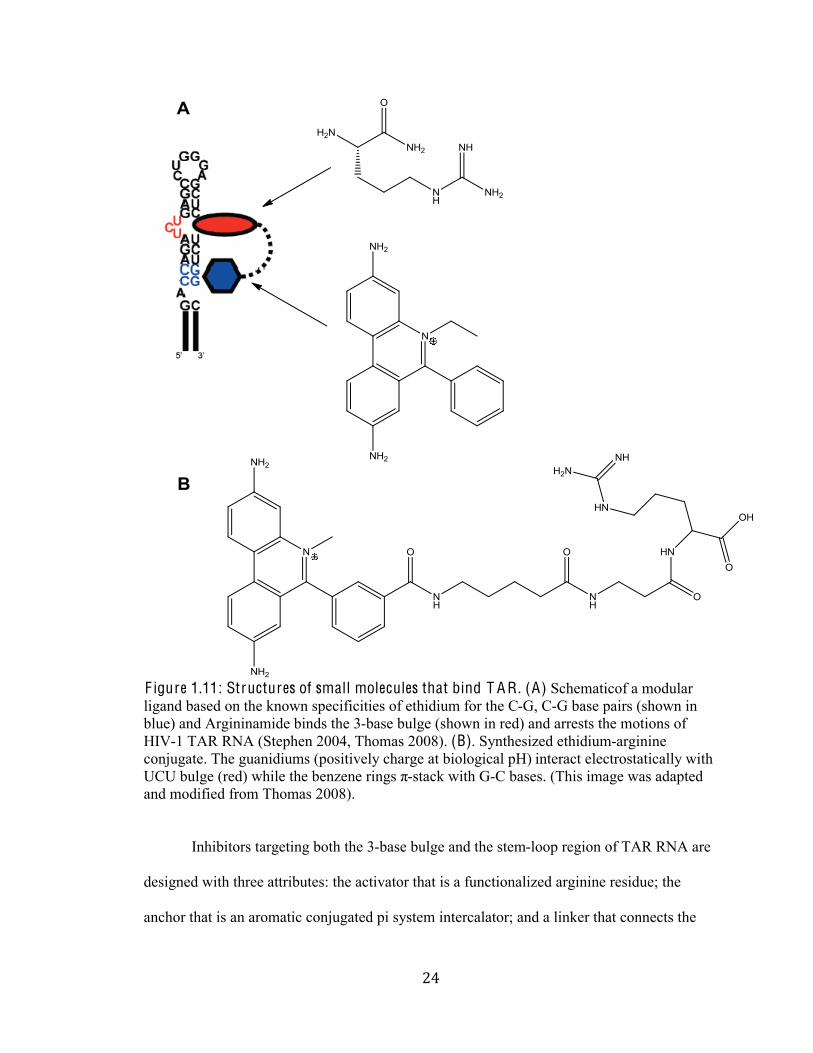
&(#
#
F igure 1.11: Structures of small molecules that bind T A R . (A) Schematicof a modular
ligand based on the known specificities of ethidium for the C-G, C-G base pairs (shown in
blue) and Argininamide binds the 3-base bulge (shown in red) and arrests the motions of
HIV-1 TAR RNA (Stephen 2004, Thomas 2008). (B). Synthesized ethidium-arginine
conjugate. The guanidiums (positively charge at biological pH) interact electrostatically with
[Q[%;*2E1%C)1:B%76(21%361%;1/\1/1%)(/E+%Z-stack with G-C bases. (This image was adapted
and modified from Thomas 2008).
Inhibitors targeting both the 3-base bulge and the stem-loop region of TAR RNA are
designed with three attributes: the activator that is a functionalized arginine residue; the
anchor that is an aromatic conjugated pi system intercalator; and a linker that connects the

&)#
#
activator and anchor (F igure 1.12). Artificial regulators with these three attributes not only
bind to TAR but also competitively block the interactions of Tat-TAR RNA.
F igure 1.12: Modular design for T A R stem-loop and 3-base-bulge inhibitors. The blue
circles (activator) represent the cationic residues of arginine or lysine responsible for the
electrostatic interactions between inhibitor and TAR-RNA. The linker connects the
intercalator (anchor) to the rest of the compound. (This image was extracted from Thomas
2008).
The aminoglycoside Neomycin-B-hexaarginine (NeoR) is a pivotal example of this type of
inhibitor (F igure 1.13). NeoR is a bi-functional inhibitor that effectively inhibits both Tat
trans-activation and Tat extracellular. NeoR accumulates in the cell nuclei and inhibits the
replication activities of HIV infected cells at concentration as low as 0.8 µM (Litovchick
2001). In addition, NeoR antagonizes Tat extracellular activities, such as increased viral
production, induction of CXCR4 expression; resulting in suppression of the HIV expression
and progression of AIDS (Litovchick 2001; Yang 2005). We believe that the potency of
NeoR is a consequence of its multivalent display of quanidiniums which increases the
flexibility of the compound. Furthermore, the fact that NeoR is a multitarget inhibitor, it is
therefore an attractive lead compound, capable of interfering with different stages of HIV
infection and AIDS pathogenesis (Litovchick 2001).
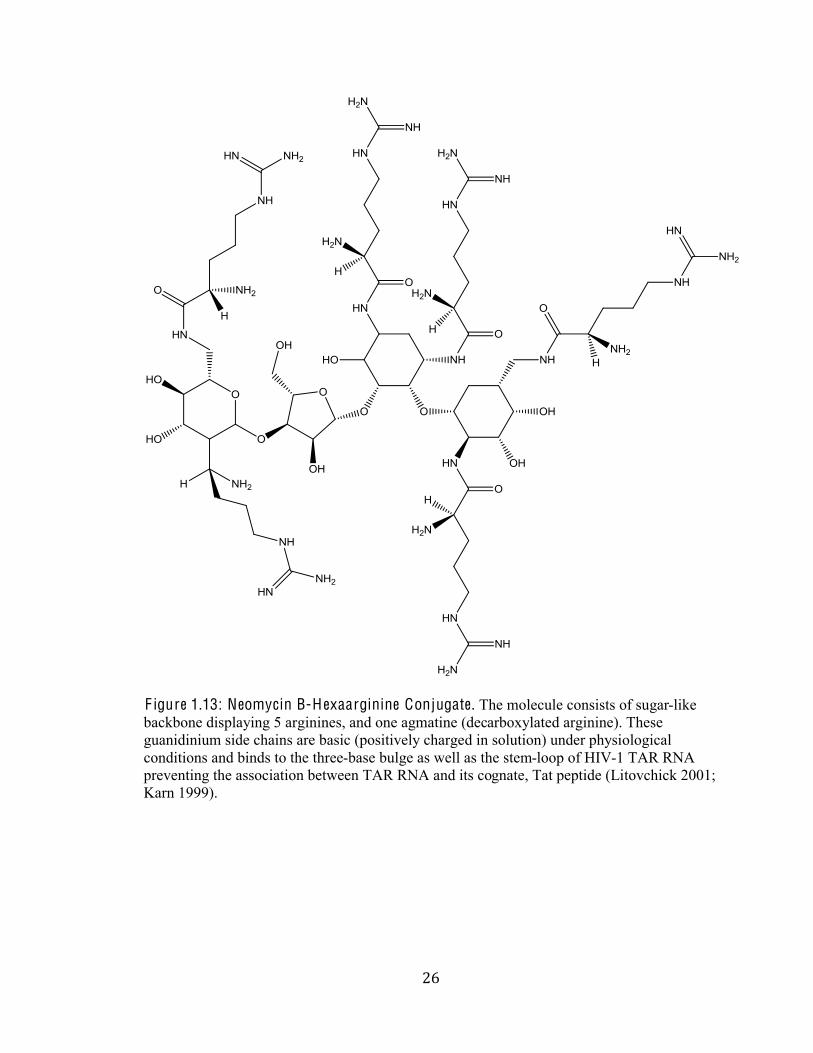
&*#
#
F igure 1.13: Neomycin B-H exaarginine Conjugate. The molecule consists of sugar-like
backbone displaying 5 arginines, and one agmatine (decarboxylated arginine). These
guanidinium side chains are basic (positively charged in solution) under physiological
conditions and binds to the three-base bulge as well as the stem-loop of HIV-1 TAR RNA
preventing the association between TAR RNA and its cognate, Tat peptide (Litovchick 2001;
Karn 1999).

&+#
#
Synthetic Polymers Targeting T A R RNA : Oligocarbamates and oligoureas were
the first synthetic peptidomimetics designed to interact with TAR RNA (F igure 1.14).
F igure 1.14: Structure of T A R RN A binding O ligocarbamate. The sequence of
oligocarbamate corresponds to Tat peptide ARM sequence 48-57
(48GlyArgLysLysArgArgGlnArgArgArg57) (Tamilarasu et al. 2001). The uses of unnatural
biopolymers containing carbamate backbone structures are attractive because they are
resistant to protease degradation in vitro. The use of the carbamate linkages (shown in red) in
this peptidomimetic increase the biological stability because they are protease resistant. (This
image was extracted from Tamilarasu et al. 2001).
Tamilarasu and co-workers found that peptidiomimetic oligomers, oligocarbamate and
oligourea are able to regulate HIV-1 gene expression both in vivo and in vitro. In HL3T1
cells, a HeLa (Human) cell line oligourea and oligocarbamate inhibit transcriptional
activation by outcompeting endogenous Tat protein with an IC50 of ~0.5 µM and 1.0 µM
respectively (Tamilarasu 2001). In addition to potency, it is important to highlight the fact
that these TAR-RNA binding oligomers are non-toxic to the human HeLa cells. The property
of non-cytotoxicity is important because molecules that are not selectively destructive to HIV
infected cells alone are potentially harmful causing adverse effects which are overall anti-
intuitive to our disease combating goals.
The most recent synthetic polymers targeting the nucleic acids of TAR RNA are
multivalent branched peptides. Branched peptides are generated from conventional peptide
synthesis using solid phase and resin techniques. However, the syntheses of the branched
forms have been shown to increases the biological stability of certain peptides in comparison

&,#
#
to the monomeric peptides. In fact, it has been shown that branched peptides are resistant to
enzyme proteolysis (Bracci 2003; Falciani 2007). In 2012, Bryson and coworkers reported
that a branched peptide, FL4 increased binding, selectivity, affinity for the three-dimensional
structure of native TAR-RNA with an half maximal inhibitory concentration (IC50) of ~1.0
µM; a consequence of multivalency (F igure 1.15) (Bryson et al. 2012). The Branched
Peptide FL4, can traverse the cell membrane of HeLa cells, and exhibit no cytotoxicity.
F igure 1.15: Schematic of combination library of branched peptides. The peptides are
synthesized on a resin via photocleavable linkage, by an automative synthesis. (This image
was extracted from Bryson 2009).
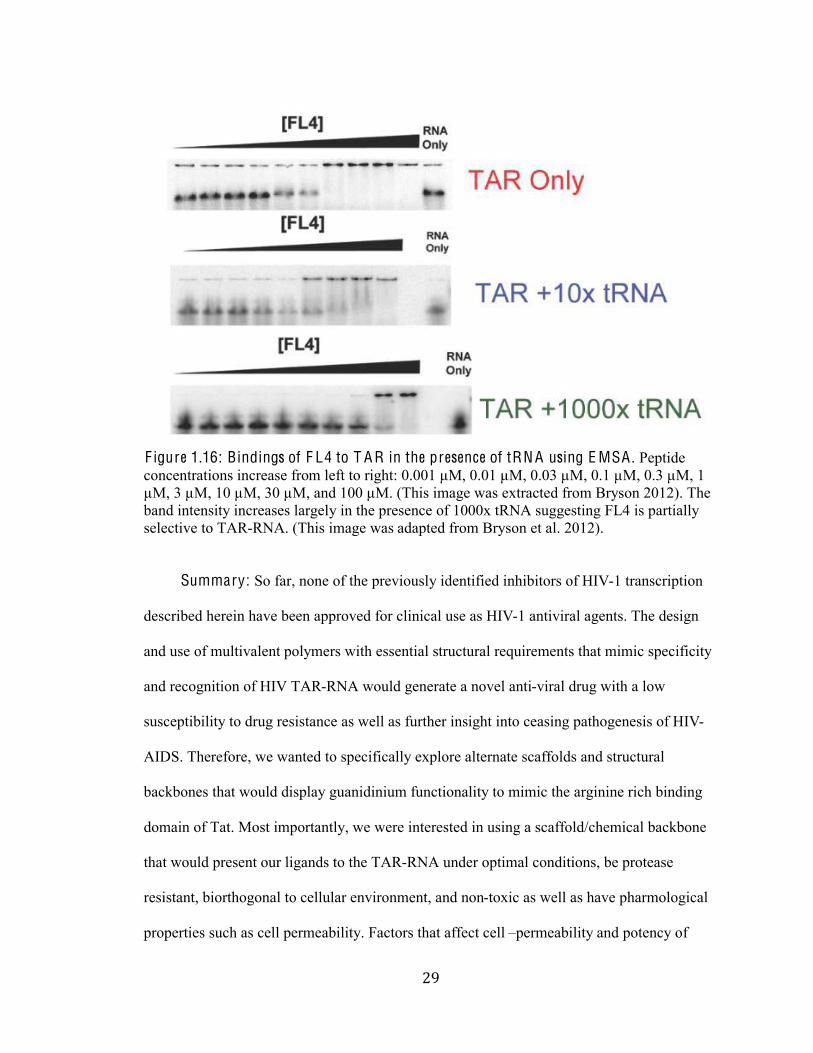
However, in competition assays between FL4 and TAR-RNA in the presence of excess tRNA
resulted in an increase shift in binding suggesting that FL4 binds with low specificity to the
TAR and therefore only partially selective for the TAR-RNA interface (F igure 2.7) (Bryson
et al. 2012). That is the branched peptide FL4 does not bind directly to the UCU bulge. It can
be hypothesized that the flexibility of the backbone may be hindering the binding affinity of
the peptide. Such hurdle can be traverse by using a more rigid structural backbone.
&-#
#
F igure 1.16: Bindings of F L4 to T A R in the presence of tRN A using E MSA . Peptide
concentrations increase from left to right: 0.001 µM, 0.01 µM, 0.03 µM, 0.1 µM, 0.3 µM, 1
µM, 3 µM, 10 µM, 30 µM, and 100 µM. (This image was extracted from Bryson 2012). The
band intensity increases largely in the presence of 1000x tRNA suggesting FL4 is partially
selective to TAR-RNA. (This image was adapted from Bryson et al. 2012).
Summary: So far, none of the previously identified inhibitors of HIV-1 transcription
described herein have been approved for clinical use as HIV-1 antiviral agents. The design
and use of multivalent polymers with essential structural requirements that mimic specificity
and recognition of HIV TAR-RNA would generate a novel anti-viral drug with a low
susceptibility to drug resistance as well as further insight into ceasing pathogenesis of HIV-
AIDS. Therefore, we wanted to specifically explore alternate scaffolds and structural
backbones that would display guanidinium functionality to mimic the arginine rich binding
domain of Tat. Most importantly, we were interested in using a scaffold/chemical backbone
that would present our ligands to the TAR-RNA under optimal conditions, be protease
resistant, biorthogonal to cellular environment, and non-toxic as well as have pharmological
properties such as cell permeability. Factors that affect cell ]permeability and potency of

'.#
#
inhibitor are molecular weight, valency (spacing) and density (number) of arginine residues
(Bryson et al. 2012).
The design of drugs to inhibit RNA-protein interactions is not a trivial task. When
developing drugs that modulate these interactions poor selectivity and binding affinity is a
common hurdle to overcome. Issues with developing synthetic molecules fall into three
categories: cost, limitations, and difficulties in synthesis including the required steps of
reaction and purification. For example, synthetic peptides normally require a significant
amount of protecting groups chemistry, several sequential coupling steps (usually in order of
de-protection, then coupling of the next ligand) and rink amide resin (solid-phase and
expensive). It is important to note that though solid-phase peptide synthesis has become
routine, the procedure presents many disadvantages. They are: the method can be laborious
and tedious; non-compatibility of resin and growing peptide chain; and formation errors in
peptides causing truncated failure sequences. These disadvantages are unfavorable owing to
the fact that peptide synthesis has proven indispensable for the structural elucidation and
activity studies of many naturally isolated products.
Thorough examination and comparison of the aforementioned compounds found to
disrupt the Tat/TAR complexation share one thing in common; their cationic nature
resembles the ARM of the Tat. These mimics are functionalized to display lysine arginine
and other guanidiniums that emulate the highly basic structural region of Tat. Therefore
prototypic, molecule designs that target TAR-RNA must be functionalized with basic
guanidiniums and amines on a carbon scaffold. In addition, while the aforementioned
analogues and mimetics are cationic in nature, the potency, affinity and specificity of the
TAR binding compounds vary in backbone structure and scaffold. Moreover, it can be
concluded that the emerging picture from these pioneering findings indicates that new types
of RNA-specific chemical scaffolds must be developed (Wang 2009).

'%#
#
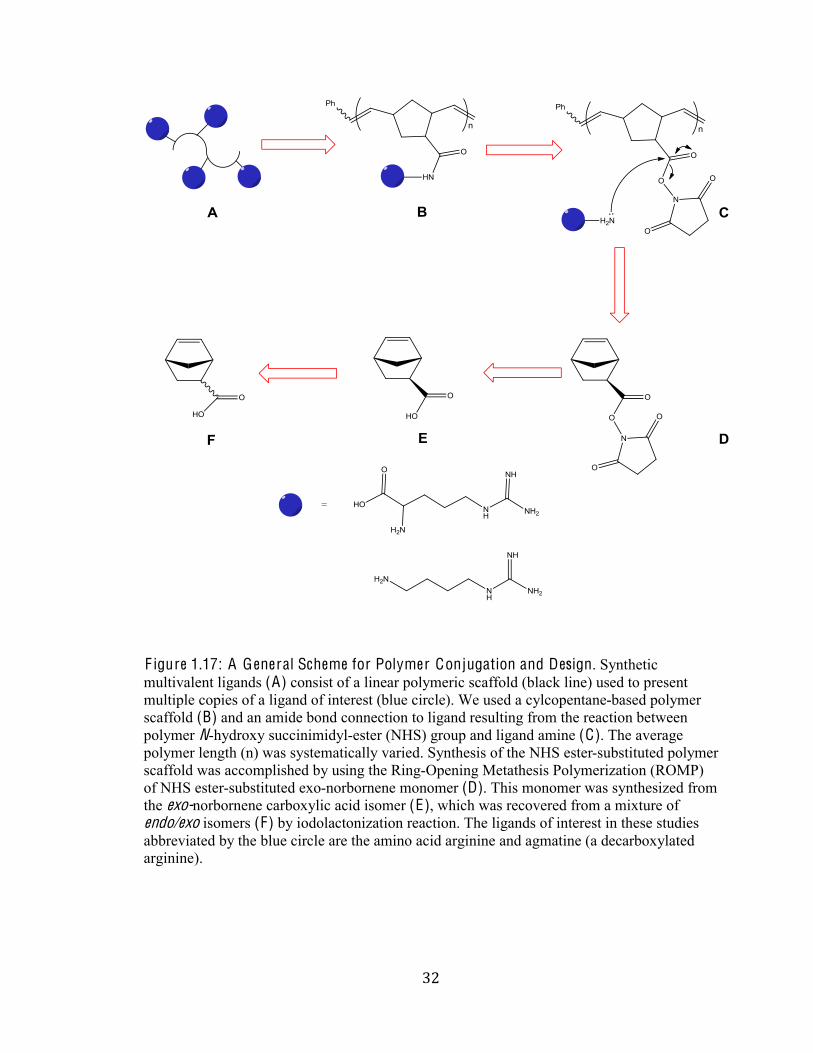
A I MS: For this project, we strategized to create polymers displaying guanidinum
functionalities. Recently, an optimized synthetic route to creating biologically active
multivalent arrays displaying desired functionality was developed. This synthesis employs
ring-opening-metathesis-polymerization (ROMP) of norbornene succinimidyl (NHS) ester
monomers (F igure 1.17) (Strong 1999). The N-hydroxysuccinimide (NHS) functionality can
be modified with desired amine-containing epitotes; because the NHS ester group is
especially sensitive to amines in the presence of other less nucleophilic groups, which makes
it ideal for coupling amino acids to the polymer units (F igure 1.17). These ROMP-derived
polymers possess an attractive featureFthe ability to control density (mole fraction) by
altering stoichiometric ratios and the control over molecular weight, a property that can have
significant influence on cellular uptake (Tamilarasu 2001; Bryson 2009). Other inherent
advantages of using multivalent ROMP derived polymers is that multivalent displays
concomitantly increase avidity and selectivity as opposed to small molecules, which possess
low selectivity and weak binding. Therefore, there is a large possibility that through
developing these multivalent polymeric displays we cannot only increase the selectivity, but
also the affinity of our polymer to the RNA or protein.
'&#
#
F igure 1.17: A General Scheme for Polymer Conjugation and Design. Synthetic
multivalent ligands (A) consist of a linear polymeric scaffold (black line) used to present
multiple copies of a ligand of interest (blue circle). We used a cylcopentane-based polymer
scaffold (B) and an amide bond connection to ligand resulting from the reaction between
polymer N-hydroxy succinimidyl-ester (NHS) group and ligand amine (C). The average
polymer length (n) was systematically varied. Synthesis of the NHS ester-substituted polymer
scaffold was accomplished by using the Ring-Opening Metathesis Polymerization (ROMP)
of NHS ester-substituted exo-norbornene monomer (D). This monomer was synthesized from
the exo-norbornene carboxylic acid isomer (E), which was recovered from a mixture of
endo/exo isomers (F) by iodolactonization reaction. The ligands of interest in these studies
abbreviated by the blue circle are the amino acid arginine and agmatine (a decarboxylated arginine).

''#
#
Synthetic Utility of R O MP-der ived Succinimidyl Ester Polymers: ROMP has been
used to develop scopes of biologically active polymers that have been used in several
structural elucidation and activity studies. In mouse B-cell activation ROMP derived
multivalent polymers were used to illicit immune responses by targeting B-cell receptors and
demonstrated no toxicity (Puffer et al. 2007), bacterial chemotaxis (Gestwicki et al. 2002)
and to develop cell permeable block copolymers (Kolonko et al. 2009). Gestwicki
demonstrated that multivalent polymers target methyl-accepting chemotaxis proteins (MCPs)
(Chemoreceptors) in bacteria subsequently influencing the signaling of cascade in bacteria.
As the valency of a galactose chemoattractant increased, a decrease in the average angular
velocity of E . coli strain AW05 (chemotaxically active E.coli) was observed, suggesting that
the bacteria were moving uniformly in response to the multivalent chemoattractant
(Gestwicki et al. 2002). Kolonko et al. also exploited this synthetic approach and length
control offered by ROMP to assemble cell permeable block copolymers (Kolonko et al.
2009). They found that block copolymers composed of half N-(3-amino propyl) guanidine
substituted and half alpha-chloroacetamide substituted were internalized into cell. The N-(3-
amino-propyl) guanidine functions as an artificial translocation domain (ATD) allowing the
polymer to traverse the cell membrane and the chloroacetamide group could be post modified
with intact proteins via reaction of cysteine side chain (Kolonko et al. 2009). Overall, they
used ROMP to generate polymeric ATD that can be used as delivery vehicles for
macromolecules as well as copolymer backbones that can promote intracellular protein
assemblies (Kolonko et al. 2009).
Determination of Protein-Nucleic Acid Interactions: Classical methods for the
detection of protein nucleic acid interactions include gel shift assays, filter-binding assays
and dot blots. However, Electrophoretic Mobility Shift Assay (EMSA) and dot blot assays
are the most popular and efficient of the three. Typical EMSA protocols, allows for solutions

'(#
#
of combined proteins and nucleic acids to be incubated and the resulting oligiomers separated
by electrophoresis under native conditions through polyacrylamide or agarose gel. Native
conditions allow for the molecules to migrate complexes of protein- RNA to migrate;
therefore complexes travel more slowly than the corresponding free nucleic acid or protein. If
the starting nucleic acid was radioisotope labeled, or fluorescently labeled nucleic acid the
gel may be analyzed using autoradiography and a fluorescent scanner respectively.
')#
#
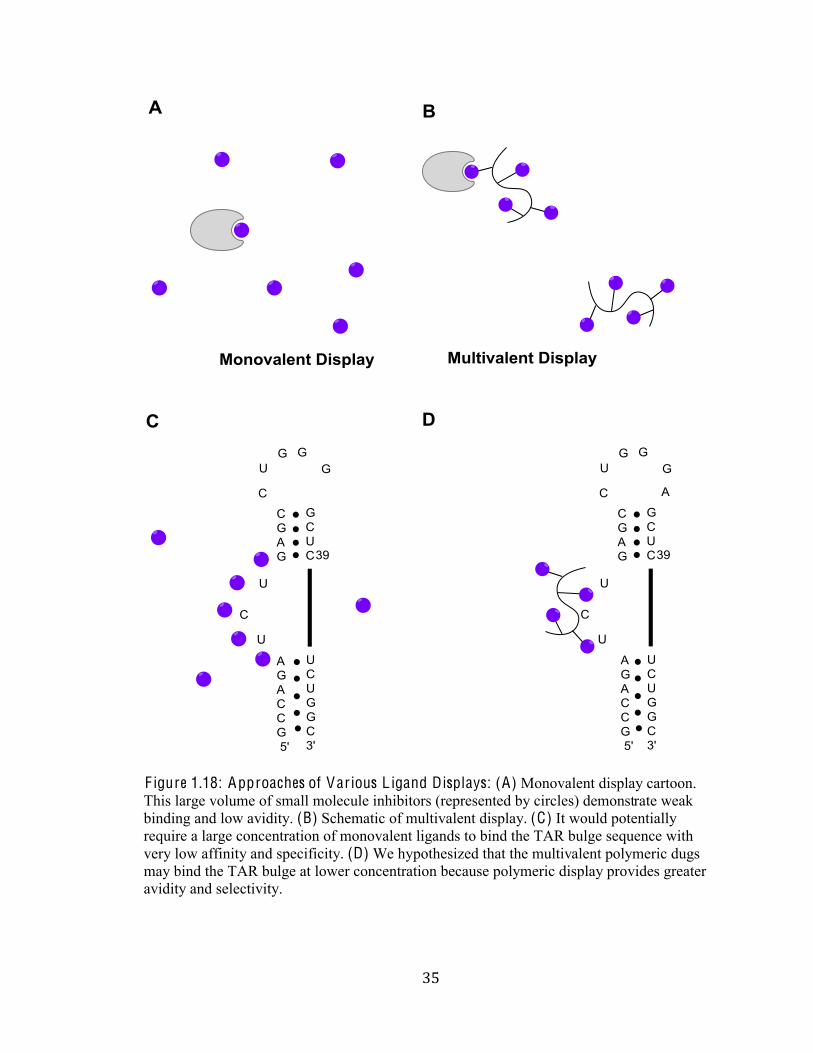
F igure 1.18: Approaches of Various L igand Displays: (A) Monovalent display cartoon.
This large volume of small molecule inhibitors (represented by circles) demonstrate weak
binding and low avidity. (B) Schematic of multivalent display. (C) It would potentially
require a large concentration of monovalent ligands to bind the TAR bulge sequence with
very low affinity and specificity. (D) We hypothesized that the multivalent polymeric dugs
may bind the TAR bulge at lower concentration because polymeric display provides greater
avidity and selectivity.

'+#
#
Materials and methods:!
Section I . Chemical Synthesis Procedures, Purification and Character ization
General Procedures and Materials. All reactions were conducted under atmospheric
conditions unless otherwise noted. All other moisture and oxygen-sensitive reactions were
performed with syringe-septum cap techniques in flame-dried glassware under an inert
nitrogen (N2) atmosphere. Reactions were carried out at room temperature unless otherwise
specified. Unless otherwise noted, all materials (reagents and solvents) were used without
further purification as obtained from commercial suppliers. Dichloromethane (CH2Cl2) was
degassed and dried by sparging with ultra-high purity argon gas followed by passage through
an activated alumina column using a Glass Contour Seca Solvent Purification System and
water (H2O) was purified with a MilliQ purification system (Millipore). Reactions were
monitored and analyzed by thin-layer chromatography (TLC) using pre-coated Silica (SiO2)
plates available from Merck. Visualization of compounds was accomplished using two
methods: staining with a potassium permanganate stain (3 g KMnO4, 20 g K2CO3, 5 mL 5%
aqueous NaOH, 300 mL H2O) and/or ultraviolet (UV) light radiation at 254 nm. Flash
Column Chromatography and disposable PD-10 desalting columns (Sephadex G-25 Medium)
was used to purify all products unless otherwise stated. Silica (SiO2) gel flash
chromatography (SiliCycle, 40-63µM, pore size 60) was used as the stationary phase for
flash chromatography, while the mobile phase varied with each procedure. Proton and carbon
Nuclear Magnetic Resonance (1H NMR and 13C NMR respectively) were obtained on a
Varian 400MHz spectrometer. 1H NMR chemical shifts were calibrated in parts per million

',#
#
(ppm) and Hertz (Hz) using residual solvents CDCl3: 1H: 7.26, 13C: 77.23; CD3OD: 1H: 3.31,
13C: 49.15; DMSO-d6: 1H: 2.5, 13C: 39.51; and D2O: 1H: 4.8. The 1HNMR spectra are
tabulated as follows: chemical shifts, multiplicities (are described using the following
abbreviations: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet and resonances
that appear broad are designated (br) numbers of protons (integration) and coupling
constants. 13C NMR spectra are reported as values in parts per million relative to residual
CHCl3 (77 ppm) or CD3OD (49 ppm) as internal standards.
Exo 5-Bicyclo[2.2.1]hepta-2- (Norbornene)-2-Carboxylic Acid (2)
A mixture of commercially available exo/endo 5-norbornene-2-
carboxylic acid 1 (predominantly endo) (5.0 mL, 40.86 mmol, 1.0
eq) was dissolved in aqueous 0.75 M sodium bicarbonate
(NaHCO3) (3.75 g/60 mL H2O). Subsequently a solution of iodine
(I2) (9.3625 g. 36.89 mmol, 0.9 eq) and potassium iodide (KI) (17.6
g, 106.2 mmol, 2.6 eq) in H2O (50 mL) (brown/black in color) was added drop-wise and
completely with a pipette. A brown-yellow color persisted at the surface, while a dark
brown/black sludge (iodolactone) was suspended at the bottom of the flask. The mixture was
decanted separating the aqueous layer from the viscous sludge. The aqueous layer was
extracted with diethyl ether (Et2O) (3 x 20 mL) to remove the residual iodolactone sludge. A
10% sodium thiosulphate (Na2S2O3) solution (0.75 g/ 6.75 mL H2O) was added to the
aqueous layer to decolorize the solution. Using a 1.0M solution of sulfuric acid H2SO4 the pH
of the solution was adjusted to two as determined by litmus paper and a yellow precipitate
was observed. The mixture was extracted again with diethyl ether (4 x 40 mL). The
combined ether layers were dried using magnesium sulfate (MgSO4) and concentrated under
reduced pressure with rotary evaporator. The concentrated solution was placed under high
vacuum for 20 minsF24 hours. The final exo acid product was typically a white/yellow solid

'-#
#
color (1.217 g, 8.807 mmol, 21.6%). 1H NMR (400 MHz, CDCl3B^%_%&&NO`%C;)%+,%&KB,%SN&S%
(dd, J = 5.9 Hz, 1H), 5.97 (dd, J = 6.3 Hz, 4.2 Hz, 1H), 3.12 (br s, 1H), 2.95 (br s, 1H), 2.30-
2.25 (ddd, J = 10.5 Hz, 4.3 Hz, 2.2 Hz, 1H), 1.96 (dt, J = 12.7 Hz, 3.6 Hz, 1H), 1.55 (d, J =
8.5 Hz, 1H), 1.45-1.38 (m, 2H).
Succinimidyl ester (N HS) substituted exo-norbornene monomer (3)
The succinimidyl ester-substituted monomer was synthesized as
reported in Strong, L.E.; Keissling. L. L. J. Am. Chem. Soc.
1999, 121, 6193-6196. The exoFnorbornene acid (1.217 g,
8.807 mmol, 1.0 eq), N-(3-dimethylaminopropyl) N-
136429-);0:((@(:1%64:)09620)(:1%Ca>Q=%b%KQ2B%C`NJc%E,%&cN`%
mmol, 1.5 eq) and N-hydroxysuccinimide (NHS) (1.521 g, 13.2
mmol, 1.5 eq) were stirred in anhydrous reaction grade CH2Cl2
(50 mL) under N2 for 24 hours. The organic layer was extracted using 5% citric acid (3 x 65
mL), saturated sodium bicarbonate NaHCO3 (2 x 50 mL) and saturated NaCl (1 x 50 mL).
The dichloromethane CH2Cl2 layer was dried with magnesium sulfate (MgSO4) and
concentrated under reduced pressure on a rotary evaporator leaving a white powdery solid.
The residue was re-dissolved in minimal amount of dichloromethane (CH2Cl2) and purified
with flash column chromatography (2:1 hexanes/ethyl acetate). A total of 60 fractions were
collected and assessed by TLC. Fractions 8 ] 58 were combined and concentrated under
reduced pressure on rotary evaporator. A white solid powder (1.197 g, 5.087 mmol, 57.8%)
was isolated and was stored in the freezer. Silica gel Rf, 0.53 (1:1 hexane/ethyl acetate); 1H
NMR (400 MHz, DMSO-d6B^%_%SN&O%C::,%&KB,%SN&`%C::, 1H), 3.26 (br s, 1H), 2.99 (br s, 1H),
2.83 (s, 4H), 2.52 (ddd, 1H), 2.05 (dt, 1H), 1.54 (d, 1H), 1.51-1.45 (m, 2H).
(.#
#
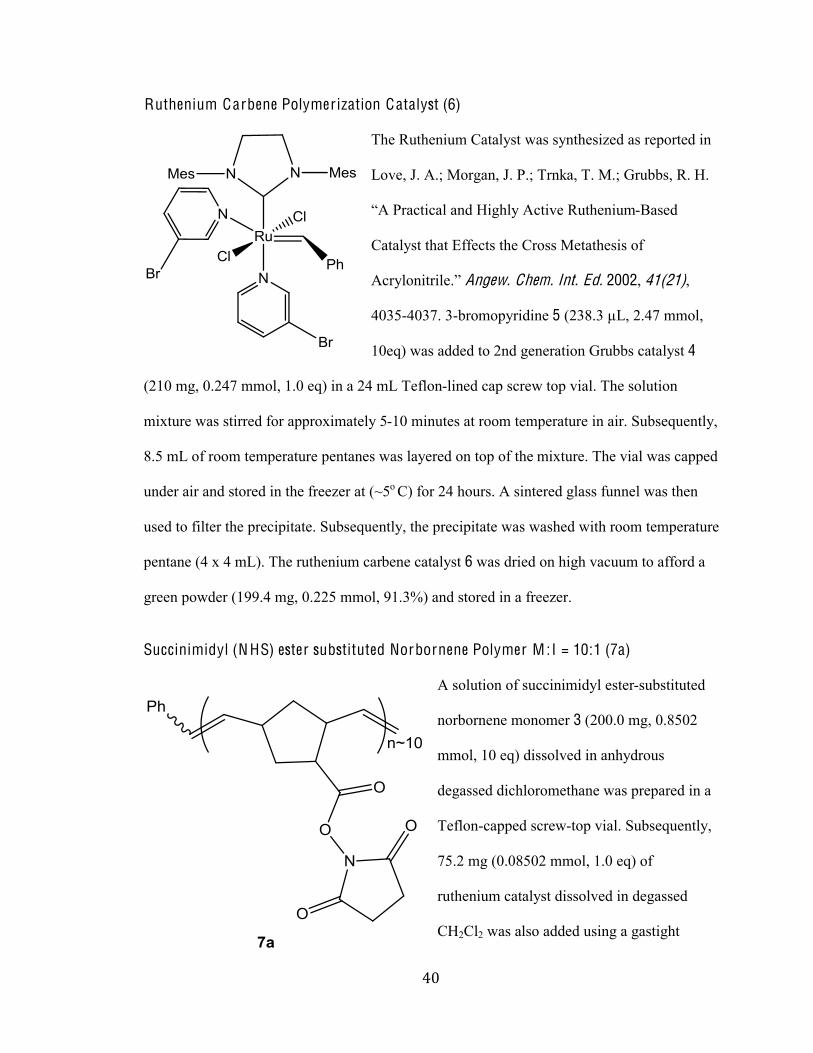
Ruthenium Carbene Polymer ization Catalyst (6)
The Ruthenium Catalyst was synthesized as reported in
Love, J. A.; Morgan, J. P.; Trnka, T. M.; Grubbs, R. H.
.<%d)-93(9-2%-/:%K(E624%<93('1%I*361/(*@-Based
Catalyst that Effects the Cross Metathesis of
<9)420/(3)(21N5%Angew. Chem. Int. Ed. 2002, 41(21),
4035-4037. 3-bromopyridine 5 (238.3 µL, 2.47 mmol,
10eq) was added to 2nd generation Grubbs catalyst 4
(210 mg, 0.247 mmol, 1.0 eq) in a 24 mL Teflon-lined cap screw top vial. The solution
mixture was stirred for approximately 5-10 minutes at room temperature in air. Subsequently,
8.5 mL of room temperature pentanes was layered on top of the mixture. The vial was capped
under air and stored in the freezer at (~5o C) for 24 hours. A sintered glass funnel was then
used to filter the precipitate. Subsequently, the precipitate was washed with room temperature
pentane (4 x 4 mL). The ruthenium carbene catalyst 6 was dried on high vacuum to afford a
green powder (199.4 mg, 0.225 mmol, 91.3%) and stored in a freezer.
Succinimidyl (N HS) ester substituted Norbornene Polymer M :I = 10:1 (7a)
A solution of succinimidyl ester-substituted
norbornene monomer 3 (200.0 mg, 0.8502
mmol, 10 eq) dissolved in anhydrous
degassed dichloromethane was prepared in a
Teflon-capped screw-top vial. Subsequently,
75.2 mg (0.08502 mmol, 1.0 eq) of
ruthenium catalyst dissolved in degassed
CH2Cl2 was also added using a gastight
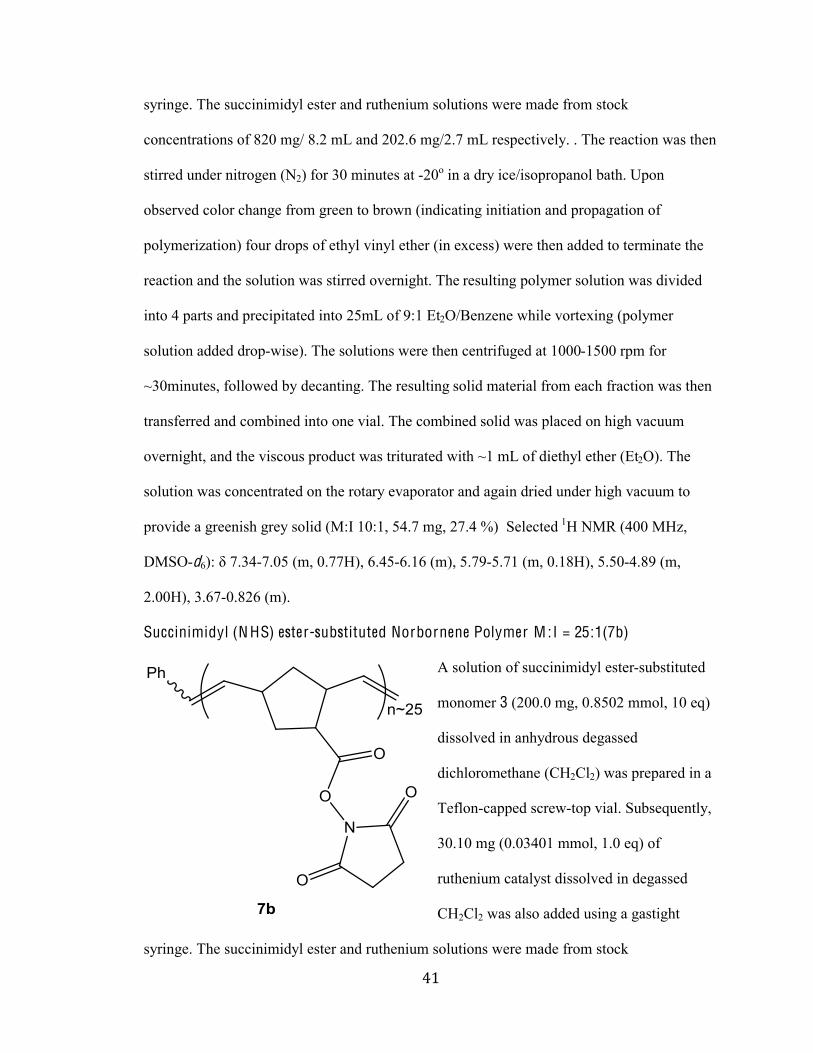
(%#
#
syringe. The succinimidyl ester and ruthenium solutions were made from stock
concentrations of 820 mg/ 8.2 mL and 202.6 mg/2.7 mL respectively. . The reaction was then
stirred under nitrogen (N2) for 30 minutes at -20o in a dry ice/isopropanol bath. Upon
observed color change from green to brown (indicating initiation and propagation of
polymerization) four drops of ethyl vinyl ether (in excess) were then added to terminate the
reaction and the solution was stirred overnight. The resulting polymer solution was divided
into 4 parts and precipitated into 25mL of 9:1 Et2O/Benzene while vortexing (polymer
solution added drop-wise). The solutions were then centrifuged at 1000-1500 rpm for
~30minutes, followed by decanting. The resulting solid material from each fraction was then
transferred and combined into one vial. The combined solid was placed on high vacuum
overnight, and the viscous product was triturated with ~1 mL of diethyl ether (Et2O). The
solution was concentrated on the rotary evaporator and again dried under high vacuum to
provide a greenish grey solid (M:I 10:1, 54.7 mg, 27.4 %) Selected 1H NMR (400 MHz,
DMSO-d6B^%_%eNcf-7.05 (m, 0.77H), 6.45-6.16 (m), 5.79-5.71 (m, 0.18H), 5.50-4.89 (m,
2.00H), 3.67-0.826 (m).
Succinimidyl (N HS) ester-substituted Norbornene Polymer M :I = 25:1(7b)
A solution of succinimidyl ester-substituted
monomer 3 (200.0 mg, 0.8502 mmol, 10 eq)
dissolved in anhydrous degassed
dichloromethane (CH2Cl2) was prepared in a
Teflon-capped screw-top vial. Subsequently,
30.10 mg (0.03401 mmol, 1.0 eq) of
ruthenium catalyst dissolved in degassed
CH2Cl2 was also added using a gastight
syringe. The succinimidyl ester and ruthenium solutions were made from stock

(&#
#
concentrations of 820 mg/ 8.2 mL and 202.6 mg/2.7 mL respectively. The reaction was then
stirred under nitrogen N2 for 30 minutes at -20 oC in a dry ice/isopropanol bath. Upon
observed color change from green to brown (indicating initiation and propagation of
polymerization) four drops of ethyl vinyl ether (in excess) were then added to terminate the
reaction and the solution was stirred overnight. The resulting polymer solution was divided
into 4 parts and precipitated into 25 mL of 9:1 Et2O/Benzene while vortexing (polymer
solution added drop-wise). The solutions were then centrifuged at 1000-1500 rpm for ~30
minutes, followed by decanting. The resulting solid material from each fraction was then
transferred and combined into one vial. The combined solid was placed on high vacuum
overnight, and the viscous product was triturated with ~1 mL of diethyl ether (Et2O). The
solution was concentrated under diminished pressure on the rotary evaporator and again dried
under high vacuum to provide a grey solid (M:I 25:1, 64.0 mg, 32 % ). Selected 1H NMR
(400 MHz, DMSO-d6B^%_%eNcf-7.05 (m, 0.40H), 6.45-6.25 (m), 6.16-5.71 (dd), 5.50-4.91 (m,
2.00H), 4.39 (s), 3.99-0.82 (m).
Succinimidyl (N HS) ester substituted Norbornene Polymer M :I = 50:1 (7c)
A solution of succinimidyl ester-substituted
norbornene monomer 3 (200.0 mg, 0.8502
mmol, 10 eq) dissolved in anhydrous
degassed dichloromethane (CH2Cl2) was
prepared in a Teflon-capped screw-top vial.
Subsequently, 15.0 mg (0.0170 mmol, 1.0 eq)
of ruthenium catalyst dissolved in degassed
CH2Cl2 was also added using a gastight
syringe. The succinimidyl ester and ruthenium solutions were made from stock
concentrations of 820 mg/ 8.2 mL and 202.6 mg/2.7 mL respectively. The reaction was then
('#
#
stirred under nitrogen N2 for 30 minutes at -20 oC in a dry ice/isopropanol bath. Upon
observed color change from green to brown (indicating initiation and propagation of
polymerization) four drops of ethyl vinyl ether (in excess) were then added to terminate the
reaction and the solution was stirred overnight. The resulting polymer solution was divided
into 4 parts and precipitated into 25 mL of 9:1 Et2O/Benzene while vortexing (polymer
solution added drop-wise). The solutions were then centrifuged at 1000-1500 rpm for ~30
minutes, followed by decanting. The resulting material from each fraction was then
transferred and combined into one vial. The combined solid was high placed on vacuum
overnight, and the viscous product was triturated with ~1 mL of diethyl ether (Et2O). The
solution was concentrated on the rotary evaporator and again dried under high vacuum to
provide a grey solid (M:I 50:1, 55.7 mg, 27.9 %). Selected 1H NMR (400 MHz, DMSO-d6):
_%eNcf-7.05 (m, 0.18H), 6.42 (m), 6.19-5.79 (dd), 5.50-4.91 (m, 2.00H), 3.98-0.468 (m).
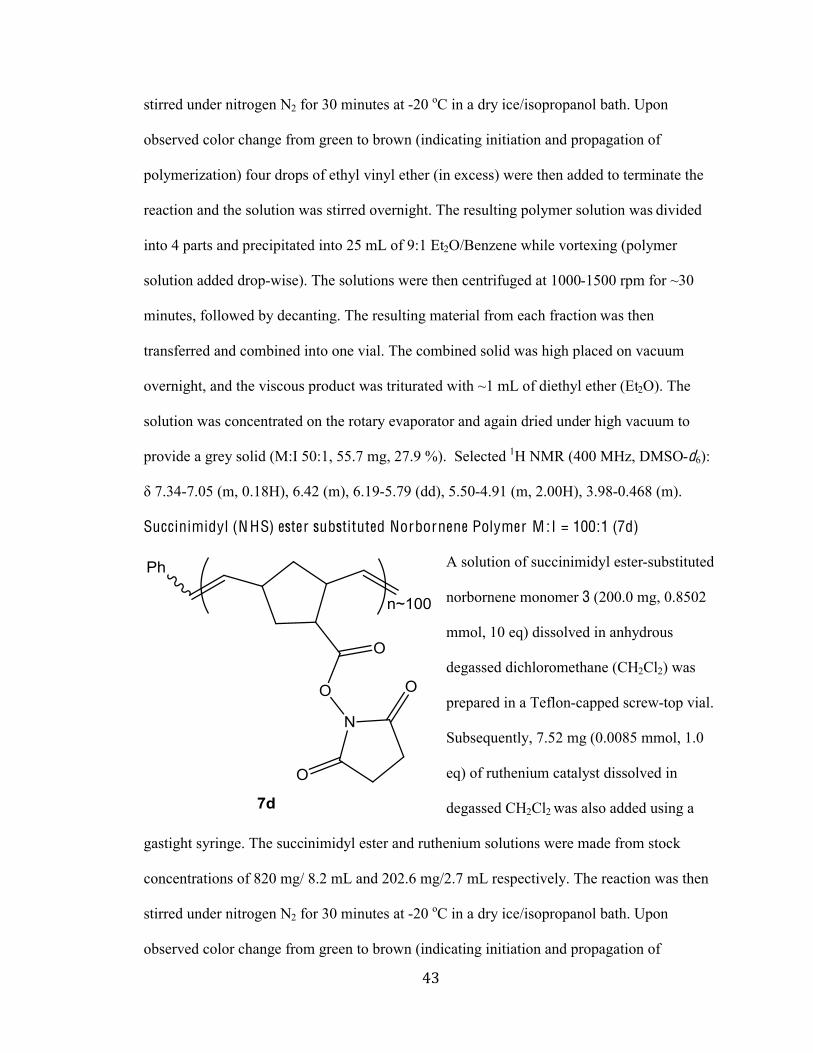
Succinimidyl (N HS) ester substituted Norbornene Polymer M :I = 100:1 (7d)
A solution of succinimidyl ester-substituted
norbornene monomer 3 (200.0 mg, 0.8502
mmol, 10 eq) dissolved in anhydrous
degassed dichloromethane (CH2Cl2) was
prepared in a Teflon-capped screw-top vial.
Subsequently, 7.52 mg (0.0085 mmol, 1.0
eq) of ruthenium catalyst dissolved in
degassed CH2Cl2 was also added using a
gastight syringe. The succinimidyl ester and ruthenium solutions were made from stock
concentrations of 820 mg/ 8.2 mL and 202.6 mg/2.7 mL respectively. The reaction was then
stirred under nitrogen N2 for 30 minutes at -20 oC in a dry ice/isopropanol bath. Upon
observed color change from green to brown (indicating initiation and propagation of

((#
#
polymerization) four drops of ethyl vinyl ether (in excess) were then added to terminate the
reaction and the solution was stirred overnight. The resulting polymer solution was divided
into 4 parts and precipitated into 25 mL of 9:1 Et2O/Benzene while vortexing (polymer
solution added drop-wise). The solution mixtures were then centrifuged at 1000-1500 rpm for
~30 minutes, followed by decanting. The resulting material from each fraction was then
transferred and combined into one vial. The combined solid was high vacuumed overnight,
and the viscous product was triturated with ~1 mL of diethyl ether (Et2O). The solution was
concentrated on the rotary evaporator and again dried under high vacuum to provide a grey
solid (M:I 100:1, 72.6 mg, 36.3 %). Selected 1H NMR (400 MHz, DMSO-d6B^%_%eNcf-7.05
(m, 0.14H), 6.19-6.16 (dd), 5.72-4.97 (m, 2.00H), 4.39 (s), 3.30-2.46 (m), 2.01-1.79 (m),
1.49-1.44 (m), 1.33-1.19 (m).
N-(2-Amino)-5-guanidinopentanoic (A rginine) Conjugated Polymer 10mer
The succinimidyl ester-substituted polymer
7a (5.3 mg, 0.0225 mmol, 1.0 eq) was
dissolved in 225 µL anhydrous DMSO. To
the solution was added 7.6 mg (0.0437
mmol, 2.0 eq) of arginine and 12 µL (0.109
mmol, 5.0 eq) of N-Methylmorpholine was
added. The reaction was vortexed to dissolve
all reagents and then left to run for three days. The reaction was quenched with 3.5 µL (2.0
eq) of 3-amino-1, 2-propanediol. Using a PD-10 desalting size exclusion column (Sephadex
G-25 resin), the product was purified. The column was first washed five times with MilliQ
water. The crude product was then loaded onto the top of the column. The reaction tube was
washed twice with 100 µL of DMSO and vortexing. The resulting solutions were loaded
onto the column as well. After polymer loaded onto the column two portions of 400 µL of

()#
#
MilliQ water was added to the top of the column and allowed to load. The column was then
filled to the top with MilliQ water and the product was eluted into 6 fractions approximately
1 mL in volume. Due to the polymers limited water solubility, the eluents containing polymer
were observed as cloudy. All the fractions were concentrated using a speed vacuum for 24
hours and weighed to provide polymer 8a. Fractions with pure solid as determined by 1H
NMR were added to solvent a system containing 490 µG%>V?gX&U%hG%>2O and thereafter
vortexed and sonicated to dissolve. The pure solid weight for 8a was 4.8 mg (M:I 10: 96 %).
The same reaction sequence was used to provide polymer 8b (M:I 25:1, 3.6 mg, 61 %) as a
flocculent grey-white solid, polymer 7c (M:I 50:1, 4. mg, 94 %) as a flocculent colorless-
solid.
Compound 8a: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.18 (br), 1.48, 1.82-2.01
(m), 2.24-2.35 (m), 2.46-2.55(m), 2.75- 2.89 (m), 3.19-3.37 (m), 4.38 (s), 4.902-4.96 (m),
5.25-5.50 (m), 7.04-7.50 (m, 5H).
Compound 8b: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.09-1.19 (br), 1.488 (br),
1.81-2.01 (d), 2.24-2.35 (m), 2.46-2.56 (m), 2.75-2.89 (m), 3.2-3.37 (m), 4.39 (s), 5.18-5.27
(d), 6.95-7.21 (m, 5H), 7.48-7.789 (m).
Compound 8c: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.02 (m), 1.19 (s), 1.47 (m),
1.82 (m), 2.29-2.35 (d), 2.44-2.59 (d), 2.62 (s), 2.89-3.00 (m), 3.21-3.28 (m), 3.39-3.46 (m),
4.43-4.66 (d), 5.13-5.27 (m), 7.6 (m, 5H)

(*#
#
N-(4-Aminobutyl) Guanidine [Agmatine] Conjugated Polymer 10 mer (9a)
The succinimidyl ester-substituted
polymer 7a (5.3 mg, 0.0225 mmol, 1.0
eq) was dissolved in 212 µL anhydrous
DMSO. To the solution 7.3 mg (0.0561
mmol, 2.5eq) of
N-(4-Aminobutyl) Guanidine
(agmatine) and11.7 µL (0.107 mmol,
5.0 eq) of N-Methylmorpholine was
added. The reaction was vortexed to dissolve all reagents and then left to run for three days.
The reaction was quenched with 3.5 µL (2.0 eq) of 3-amino-1, 2-propanediol. Using a PD-10
desalting size exclusion column (Sephadex G-25 resin), the product was purified. The
column was first washed five times with MilliQ water. The crude product was then loaded
onto the top of the column. The reaction tube was washed twice with 100 µL of DMSO and
vortexing. The resulting solutions were loaded onto the column as well. After polymer
loaded onto the column two portions of 400 µL of MilliQ water was added to the top of the
column and allowed to load. The column was then filled to the top with MilliQ water and the
product was eluted into 5 fractions approximately 1 mL in volume. Due to the polymers
limited water solubility, the eluents containing polymer were observed as cloudy. All the
fractions were concentrated using a speed vacuum for 24 hours to evaporate the water and
weighed (F1 0.0001 mg, F2 0.0035 mg, F3 0.0002 mg, F4 0.00 mg, F5 0.00 mg). Fractions
with pure solid as determined by 1H NMR were added to solvent a system containing 490 µL
>V?gX&U%hG%>2O and thereafter vortexed and sonicated to dissolve. The pure solid weight
for 9a was 3.8 mg (M:I 10:1,76% ). The same reaction sequence was used to provide
polymer 9b (M:I 25:1, 4.2 mg, 84% ) as a flocculent brown solid, polymer 9c (M:I 50:1, 4.1
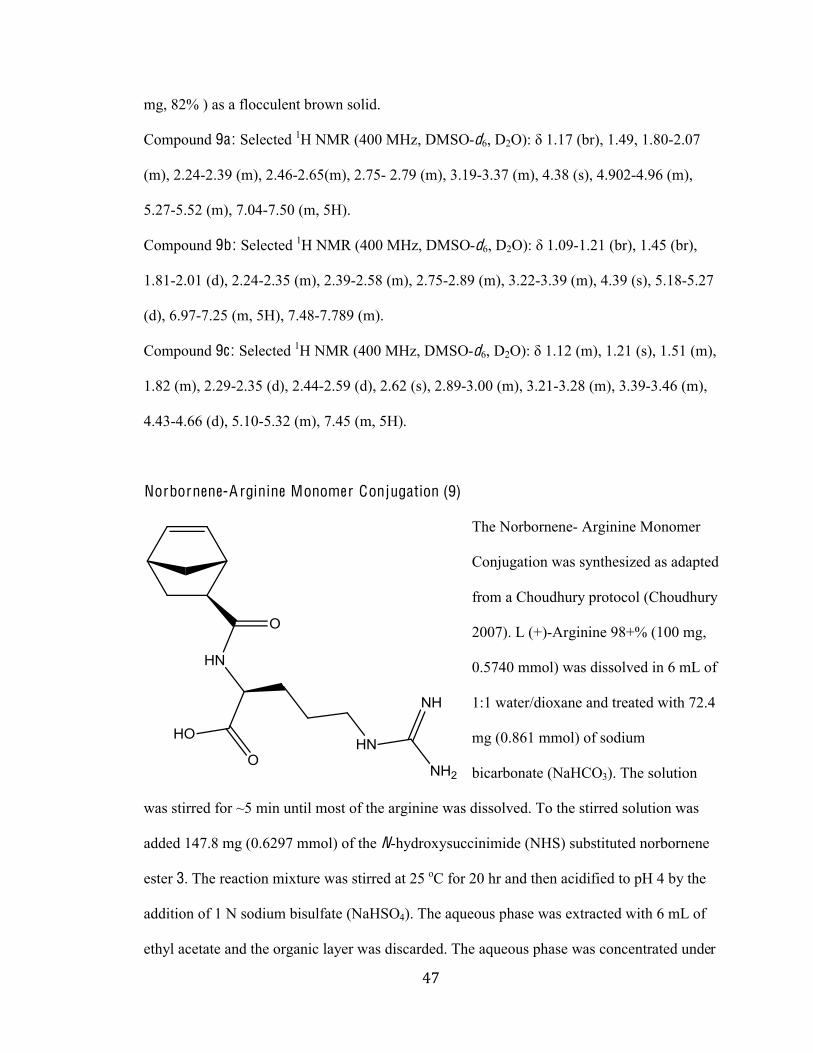
(+#
#
mg, 82% ) as a flocculent brown solid.
Compound 9a: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.17 (br), 1.49, 1.80-2.07
(m), 2.24-2.39 (m), 2.46-2.65(m), 2.75- 2.79 (m), 3.19-3.37 (m), 4.38 (s), 4.902-4.96 (m),
5.27-5.52 (m), 7.04-7.50 (m, 5H).
Compound 9b: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.09-1.21 (br), 1.45 (br),
1.81-2.01 (d), 2.24-2.35 (m), 2.39-2.58 (m), 2.75-2.89 (m), 3.22-3.39 (m), 4.39 (s), 5.18-5.27
(d), 6.97-7.25 (m, 5H), 7.48-7.789 (m).
Compound 9c: Selected 1H NMR (400 MHz, DMSO-d6, D2gB^%_ 1.12 (m), 1.21 (s), 1.51 (m),
1.82 (m), 2.29-2.35 (d), 2.44-2.59 (d), 2.62 (s), 2.89-3.00 (m), 3.21-3.28 (m), 3.39-3.46 (m),
4.43-4.66 (d), 5.10-5.32 (m), 7.45 (m, 5H).
Norbornene-A rginine Monomer Conjugation (9)
The Norbornene- Arginine Monomer
Conjugation was synthesized as adapted
from a Choudhury protocol (Choudhury
2007). L (+)-Arginine 98+% (100 mg,
0.5740 mmol) was dissolved in 6 mL of
1:1 water/dioxane and treated with 72.4
mg (0.861 mmol) of sodium
bicarbonate (NaHCO3). The solution
was stirred for ~5 min until most of the arginine was dissolved. To the stirred solution was
added 147.8 mg (0.6297 mmol) of the N-hydroxysuccinimide (NHS) substituted norbornene
ester 3. The reaction mixture was stirred at 25 oC for 20 hr and then acidified to pH 4 by the
addition of 1 N sodium bisulfate (NaHSO4). The aqueous phase was extracted with 6 mL of
ethyl acetate and the organic layer was discarded. The aqueous phase was concentrated under

(,#
#
reduced pressure. The residue was suspended in 4 mL of methanol and the insoluble solid
was vacuum- filtered using a frit funnel. Subsequently, the solid was washed twice with 3mL
portions of methanol. The combined methanol extracts were concentrated under reduced
pressure. The vicious oil residue was further purified by flash chromatography on a silica gel
column. Elution with 15mL 1:1, 2:1, and then 3:1 methanol-ethyl acetate afforded ideal
separation. There were 25 fractions in total and the pure fractions 2-20 were qualitatively
assessed by thin-layer-chromatography and thereafter combined and concentrated on the
rotary evaporator. The viscous residual was dried under high vacuum to afford the Arginine-
substituted monomer 9 as a colorless foam: yield 188 mg (96 %); silica gel TLC Rf , 0.41 (4:1
methanol-ethyl acetate); 1K%!VI%CfUU%VK\,%>V?gB^%_%%&UNPJ%C;)%+,%&KB,%PNef%C;)%+,%`KB,%
7.67 (br d, 1H), 6.08 (dd, 1H), 6.04 (dd, 1H), 5.8 (d, 1H), 4.01 (dt, 1H), 2.78 (m, 2H), 2.29 (d,
1H), 1.95-1.15 (m, 10H).

(-#
#
Section I I : Biological Assays
Mater ials and Reagents:
O ligonucleotide. Non-labelled wild-type TAR RNA (MW = 9, 290.6 g/mole OD 260=3.1,
&&NJ%/@021+,%UN&&%@E,%+1i*1/91%J"-rGrCrC rArGrA rUrCrU rGrArG rCrCrU rGrGrG
rArGrC rUrCrU rCrUrG rGrC -c"B%7-+%-9i*()1:%A)0@%=/31E)-31:%H196/020E(1+%C=>H,%
Coralville, I). The extinction coefficient was 268, 900 L/(mol cm) at absorbance peak of 260
nm. RNase-Free HPLC by manufacturer purified TAR RNA and no further purification was
administered. 2.0 mg of Tat (47- 58) peptide (NH2-YGRKKRRQRRRP-COOH) (with
OONfUj%KdGQ%8*)(34T%VR%k%&SJeNOe%EX@02B%7-+%0;3-(/1:%A)0@%Z-Proteomics, LLC
(Huntsville, AL).
Molecular Biology Kits:
Electrophoretic Mobility-Shift Assay (EMSA) Kit was obtained from Invitrogen (Grand
Island, NY) and consisted of: SYBR® Green EMSA nucleic acid gel stain (Component A),
&UU%hG%0A%-%&U,UUUl%90/91/3)-31%(/%:(@13642+*2A0m(:1%C>V?gB,%?ndIgo%I*;4%aV?<%
protein gel stain (Component B), 650 mL of an aqueous 1X solution, Trichloroacetic acid
(TCA, Component C), 87.5 g, 6X EMSA gel-loading solution (Component D), 1 mL and 5X
;(/:(/E%;*AA1)%CQ0@80/1/3%aB,%`UU%hGN%
EMSA is a robust technique that provides a fast, easy and quantitative method to detect both
nucleic acid and protein in the same gel. This kit uses two fluorescent dyes for detection -
SYBR® Green EMSA nucleic acid gel stain for RNA or DNA detection and SYPRO® Ruby
EMSA protein gel stain for protein detection. The nucleic acids and proteins are stained
sequentially in the gel after electrophoresis, and labeling does not interfere with the protein
binding being assayed. All the reagents were used without further purification and diluted to

).#
#
achieve selective staining conditions.
Native PAGE Reagents:
Native polyacrylamide 16 % (w/v) PAGE gel cassettes (8 x 10) cm, Tris-Tricine based
RunBlue Native Run Buffer (20X), and RunBlue Native Sample Buffer (4X) were obtained
commercially from Expedeon. All the Native PAGE reagents were used as acquired and
diluted accordingly for each reaction condition.
Silver Staining Reagents:
Silver Nitrate ACS Grade and Formaldehyde, 37% Solution Proteomics Grade was obtained
from VWR (Amresco; Solon, OH, Cat# 97064-582 and Cat# 97064-886 respectively).
Sodium Thiosulfate, 0.0250 Normal (N/40) was obtained from VWR (Ricca; Cat#
RC790032). Absolute Ethanol (EtOH) and Acetic acid (HOAc)
was obtained from Sigma Aldrich (St. Louis, MO). Sodium bicarbonate was also obtained
from VWR (Amresco; Solon, OH).
Solutions:
DEPC-Treated Water:
Diethyl pyrocarbonate (DEPC)-treated water was prepared to free deionized water of
possible contaminating RNases. To every 1000 mL of deionized water approximately 1 mL
of 0.1% diethyl pyrocarbonate was added to the solution and mixed vigorously at ambient
temperature for 1 hr. The solution was then autoclaved from 45 minutes to inactivate DEPC
by hydrolyzing diethylpyrocarbonate to carbon dioxide and ethanol. These byproducts are
volatile so are released as vapors yielding RNase free water. The solution is then cooled to
room temperature prior to use.

)%#
#
TE (Tris-EDTA) Buffer Solution:
Stock buffer solutions of Tris/EDTA (Ethylenediamine Tetraacetic Acid) were prepared for
diluting TAR RNA samples. The TE solutions were made of
Tris(hydroxymethyl)aminoethane base (Tris-base), EDTA, HCl in DEPC treated water. In
each solution, final buffer concentrations were 10 mM (604.8 mg/0.5 L) Tris-base, and 0.5
mM (9.3 mg/0.5 L) EDTA at ~pH 8.0. The buffer was further sterilized by filtration through
a 0.22-µM filter.
Magnesium Binding Buffer Stock Solution:
A stock binding buffer solution of 100 mM Tris-HCl (606.8 mg/50 mL), 250 mM KCl (93.2
mg/50 mL), 5 mM MgCl2 (52.3 mg/50 mL), 5 mM dithiothreitol (DTT) (39.0 mg/ 50 mL)
and 25% Glycerin (12.5 mL/50 mL) was prepared using DEPC-treated water.
Silver Staining Solutions:
0.2% Silver nitrate:
0.2 g AgNO3
100 mL ddH2O
0.05% Formaldehyde:
50 µL of 37% formaldehyde
100mL of ddH2O
30% Ethanol:
300 mL of Absolute Ethanol
1 Liter of ddH2O
10% Acetic Acids:
50 mL of Glacial Acetic Acid
500 mL of ddH2O

)&#
#
0.08% Formaldehyde:
80 µL of 37% formaldehyde
100 mL of ddH2O
2.5 mM Sodium Thiosulfate:
0.31 g sodium thiosulfate (Na2S2O3)
500 mL ddH2O
3% Sodium Bicarbonate:
7.5 g Na2CO3
250 mL dd H2O
`U%hV%?0:(*@%H6(0+*2A-31^%
800 µL of 2.5 mM Na2S2O3
100mL ddH2O
Methods:
Preparation of Samples for Electrophoretic Mobility-Shift Assay (EMSA):
HIV-1 TAR RNA (0.11 mg, 11.5 nmole) was diluted with 115 µL of TE buffer to make a
final stock concent)-3(0/%0A%&UU%hV%C&UUUlBN%H1/-fold serial dilutions were made to generate
stock concentrations of 10 µM (100X), 1 µM (10X), and 100 nM (1X).
H70%@(22(E)-@+%0A%H-3%8183(:1%7-+%:(++02'1:%(/%&`U%hG%0A%Ha%;*AA1)%30%@-p1%-%A(/-2%+02*3(0/%
stock concentration of 10.05 mM. Additional ten-fold serial dilutions were performed to
E1/1)-31%+309p%90/91/3)-3(0/+%0A%&%@V,%&UU%hV,%&U%hV,%-/:%&%hVN
Preparations of dilutions for Electrophoretic mobility Shift Assay:
<%UN&%@Vk&UU%hV%+309p%+02*3(0/%0A%/*921(9%-9(:%7-+%8)1pared by dissolving (11.5nmoles,
UN&&@EB%0A%H<I%I!<%(/%&&J%hG%0A%A)1+624%8)18-)1:%Ha%;*AA1)%C&U@V%H)(+-HCL ~pH 8.0,
1mM EDTA) and gently vortexed to create and homogenous mixture. To decrease the
amount of freeze thaw cycles of the original stock of TAR RNA serial dilutions of 1:10,
1:100 and 1:1000 were prepared. A 1:10 dilution from this stock was by diluted by adding 10

)'#
#
hG%0A%&UU%hV%H<I%+309p%-/:%:(++02'1:%(/%OU%hG%0A%Ha%;*AA1)%(/%-%207%-:61+(0/%1881/:0)A%30%
9)1-31%-%&U%hV%+309p%+02*3(0/%-/:%+7()21:%30%@ake solution homogenous. The 1:100 dilutions
7-+%@-:1%;4%-::(/E%&U%hG%0A%361%&U%hV%+02*3(0/+%30%-/0361)%OU%hG%0A%Ha%;*AA1)%30%E1/1)-31%
-%&%hV%+02*3(0/%76(96%7-+%A*)361)%:(2*31:%;4%+-@1%90/'1/3(0/%30%9)1-31%-%&^&UUU%
dilution/100 nM stock solution.
Concerns with TAR RNA sample quality, and the eventual failure of resolving bands
on the gel prompted spectrophotometric analysis using Nano Drop (ND-1000) UV-Vis
Spectrophotometer. The spectral analysis data of the HIV-1 stock and diluted samples
revealed ideal A260/280 ratios of 2.1.
General Procedure for TAR RNA Control Assays:
Five microliter aliquots of TAR RNA in TE buffer (10 mM Tris-HCl, 0.5 mM
EDTA) was then added to an empty low-adhesion eppendorf with RNase free low-adhesion
pipettes. In addition, 3 hG%0A%::%K2g%-/:%`%hG%0A%Jl%;(/:(/E%;*AA1)%C750 mM KCl, 0.5 mM
dithiothreitol, 0.5 mM EDTA, 50 mM Tris, pH 7.4). To make sure the control environments
of the TAR RNA resembled that of the forthcoming inhibition assay the TAR RNA was
incubated in 5 mL a 49:1 of DMSO/D2g%+02*3(0/%9)1-3(/E%-%A(/-2%)1-93(0/%'02*@1%0A%&J%hG%
and mixed gently but thoroughly. The reaction minutes was then incubated at 37 oC for
approximately 30 minutes.
General Procedure for Inhibition Assays without Magnesium Binding Buffer:
Initially, the binding of our ROMP-derived polymeric guanidiniums to the TAR RNA
interactions were studied by EMSA performed on 16% w/v Native polyacrylamide gels in
low ionic strength commercially available Native Run Buffer (Tricine, Tris-
hydromethylaminomethane, sodium bisulfite). The polymeric guanidiniums were diluted in

)(#
#
fOU%hG%>V?gX%&U%hG%>2O to prepare each series at 2X concentrations. Seven and half
microliter aliquots of HIV-&%H<I%I!<%(/%Ha%;*AA1)%71)1%-::1:%30%eNJ%hG%-2(i*03+%0A%361%
conjugated multivalent polymer to give the desired final concentration of each component.
The binding of ROMP-derived multivalent guanidiniums to the TAR RNA was assayed by
incubating the commercially acquired wild type TAR RNA with increasing concentrations of
the specific-polymer lengths for approximately 30 minutes at 37 oQ%(/%361%8)1+1/91%0A%c%hG%
6X binding buffer (50 mM Tris-HCl (pH 7.4 at 20), 750 mM KCl, 0.5 mM EDTA, 0.5 mM
:(36(036)1(302BN%H61)1-A31),%J%hG%0A%-%fl%aV?<%E12-loading buffer solution was added to the
mixture for loading.
General Procedure for Inhibition Assays in the presence of Magnesium Binding Buffer:
Five microliter aliquots of HIV-1 TAR RNA in TE buffer were pre-(/9*;-31:%7(36%J%hG%
aliquots of magnesium binding buffer (20 mM Tris-HCL, 50 mM KCL, 1 mM MgCl2, 5 mM
DTT, 5% Gylcerin) for approximately 30 minutes at ambient temperature. Following pre-
(/9*;-3(0/,%361%H<I%I!<%7-+%3(3)-31:%7(36%J%hG%-2(i*03+%0A%361%-88)08)(-31%90/Y*E-31:%
multivalent polymer to give the desired final concentration of each component. The final
concentration of TAR RNA was 200nM while the concentration of specific polymer
(/9)1-+1:%-+%A02207+^%&%hV,%J%hV,%&U%hVN%%H61%303-2%'02*@1%@(m3*)1%0A%A(A311/%@(9)02(31)%7-+%
then incubated for 37 oQ%A0)%cU%@(/*31+N%H61)1-A31),%J%hG%0A%-%fl%aV?<%E12-loading buffer
solution was added to the mixture for loading.
Native-PAGE Gels Electrophoresis of TAR RNA control samples:
q02207(/E%361%-88)08)(-31%(/9*;-3(0/%0A%H<I%I!<%`NJU%hG%0A%Sl%aV?<%E12%20-:(/E%
solution was added and mixed gently. Before loading samples, the wells very rinse twice
thoroughly with ultrapure water. Thin RNase free, and low adhesion pipette tips were then

))#
#
used to load 12-&J%hG%0A%1-96%+-@821%/1-)%361%;0330@%0A%361%7122N%The RNA was then
electrophoresed on the 16 % non-denaturing polyacrylamide gels for 90 minutes at 130 V,
4oC.
TAR RNA SYBR® Green EMSA Nucleic Acid Staining:
The protocol used for the nucleic acid staining of TAR RNA was adapted from
Invitrogen product fact sheet. For a typical gel staining the SYBR® Green EMSA stain is
diluted into of TE (10 mM Tris-HCl, 1mM EDTA, pH~8.0). A total of 5 µL the 10 000X
stain concentrate is diluted into 50 mL of TE buffer generating a 1X final concentration. The
electrophoresed gel was then place in a clean Rubbermaid Servin Saver plastic staining
container followed by addition of sufficient 1X SYBR Green stain. The gel was then
incubated in the 1X stain covered with aluminum foil to protect from light with continuous
agitation on an orbital shaker at 50 rpm from ~40 minutes at ambient temperature (25 oC).
Note that each 50 mL staining solution was kept in the cold room at 4 oC in the dark and
reused for a total of three-four times. Following this, the gel was washed twice with 150 mL
of RNase free dH2O for ~10 seconds to remove excess stain.
Silver Staining:
The protocol used for staining the Protein was adapted from a Chevallet protocol
(Chevallet 2006) and in the laboratory of Melissa Kosinski-Collins, under the supervision of
Deborah Bordne.
Following electrophoresis the gel was incubated in a fixer solution of 30% ethanol
(EtOH) and 10% acetic acid (HOAc) for 2 hrs. Subsequently, the gel was washed with 30%
EtOH twice every 15 minutes for 30 minutes and thereafter rehydrated by washing with dd
H2O for approximately 25 minutes to remove all the acetic acid, reduce background staining

)*#
#
and increase the gel sensitivity. The gel was then sensitized by soaking in 0.02%, (2.5 mM)
sodium thiosulfate (Na2S2O3) for 2 minutes and washed with ddH2O two times every 5
minutes for 10 minutes. Afterwards, the gel was impregnated with silver nitrate by incubating
for 30 minutes in at 4o C cold. Following this, the gel was then washed with ddH2O twice in 1
min. The gel was then placed in a new staining tray and developed in 3% sodium bicarbonate
(Na2CO3B,%UNUJj%A0)@-2:164:1%CJU%hG%cej%QK2gB,%`U%hV%+0:(*@%36(0+*2A-31%C!-2S2O3).
The developer solution was changed immediately when a yellow color change was observed.
The process was terminated when staining was sufficient. The gel staining was terminated by
washed with ddH2O for 30 seconds and incubation in 10 mL of acetic acid (HOAc) for five
minutes. Lastly, the gel was then left at 4 ºC in 1.0 % HOAc for storage. Prior to analysis the
gel was washed in ddH2O for 3 x 10 min to ensure complete removal of acetic acid.
Staining with SYPRO® Ruby EMSA protein gel stain:
The protocol for staining Tat protein with SYPRO® Ruby stain was adapted from
Invitrogen fact sheet and in the laboratory of Melissa-Kosinski Collins.
To a solution a bottle containing 87.5 g of Trichloroacetic (TCA) a 100 mL of
SYPRO® Ruby EMSA protein gel stain was added completely. The solution was agitated
and shaken to dissolve the TCA. The gel was then placed in a clean Rubbermaid Servin
Saver plastic staining container followed by addition of 50 mL SYPRO® Ruby EMSA
protein gel stain in TCA. The gel was incubated with continuous gentle agitation on an
orbital shaker at 50 rpm for approximately 3 hours in the dark. Subsequently, the gel was
washed twice with 150 mL of dd H2O for about 10 seconds. The gel was further de-stained
with 10 % methanol and 7% acetic acid for 60 minutes. The gel was again washed in twice
with 150 mL of dd H2O to remove all the acetic acid.

)+#
#
Data Acquisition, Processing and Analysis:
Electrophoresis data were collected on Molecular Imager Gel Doc XR acquired from
Bio-Rad Laboratories Inc., a Typhoon Scanner (Amersham Biosciences) and Image
Quantification Software (Armersham Biosciences). Xcel graphs were further generated to
tabulate the quantities of binding.

),#
#
Results: Section I : Synthesis of Guanidinylated-Substituted Polymers The first major goal of our research project was to synthesized multivalent
peptidomimetics with enhanced inhibitor potency, specificity and affinity for interaction with
the 3-base-bulge interface of tertiary folding TAR-RNA. Through synthetic rationale
garnered from researching previously reported TAR-RNA inhibitors, we aim to generate
multivalent oligomers of defined lengths and low polydispersities using post-polymerization
modification strategy to conjugate ligands onto a polymeric scaffold (Strong 1999). Post-
synthetic ligand modification would allow us to attain the structural requirements for
inhibiting TAR RNA/Tat association. Our efforts will begin by synthesizing monomers
containing amine-reactive succinimidyl ester groups. Subsequently, the polymeric scaffolds
of well-defined lengths will be generated via ring-opening-metathesis polymerization
(ROMP) of the amine-reactive monomers. ROMP is an attractive method for polymerization
because it utilizes a reactive ruthenium carbine catalyst, which can tailor polymers of well-
defined lengths by varying the monomer to catalyst initiator ratios (Strong 1999; Puffer 2007;
Gestwicki 2002).
Norbornene Monomers: Synthesis of the isomerically pure exo- norbornene
monomer (2) for ROMP was generated in one step (Scheme 1). An iodolactonization reaction
of a commercially available mixture of exo and endo stereoisomers of 5-norbornene-2-
carboxylic acid (1) was performed.

)-#
#
Scheme 1: Isolation of Exo-Norbornene. Iodolactonization of a commercially
available mixture of endo/exonorbornene carboxylic acid (1) afforded the exo isomer
(2). Reagents and conditions: Predominantly endo/exo-norbornene acid (1) (5 mL,
40 mmol, 1.0 eq), I2 (9.37 g, 36.9 mmol, 0.87 eq), KI (17.6 g, 106.2 mmol, 2.6 eq),
and NaHCO3 (3.75 g/60 mL), H2O, 25o C, 4 h.
The general mechanism of the iodolactonization is detailed in F igure 3.1. The predominantly
endo mixture was dissolved in aqueous sodium bicarbonate (NaHCO3) and treated with an
amalgamation of iodine (I2) and potassium iodide (KI). The endo isomer (1 endo), which
reacts very quickly with iodine, is precipitated as iodolactone, a brown sludge, while the exo
isomer reacts slowly to form a di-iodide compound (F igure 3.1). Because the diiodide is
unstable at room temperature it decomposes readily back to the alkene and the iodine.
Subsequently, the exo product was isolated via extraction with diethyl ether; the iodolactone
dissolved in the ether layer was discarded, while the exo-isomer remained in aqueous layer.
Acidification of the aqueous solution with sodium thiosulfate removed excess iodine and
afforded the 2 exo product. It was necessary to isolate the 2 because mechanistic studies have
shown that the exo iosomer reacts faster with the ROMP catalyst. Furthermore, the endo
conformation will sterically and electronically inhibit polymerization in downstream
reactions as a result of: (1) unfavorable steric interactions between the growing polymer
chains and the incoming isomer and (2) the orientation and electronics of the carbonyl group
additionally plays a critical role in retarding the rate of propagation. Overall, the exo isomer
is necessary because, this configuration allows for greater reactivity under ring-opening-
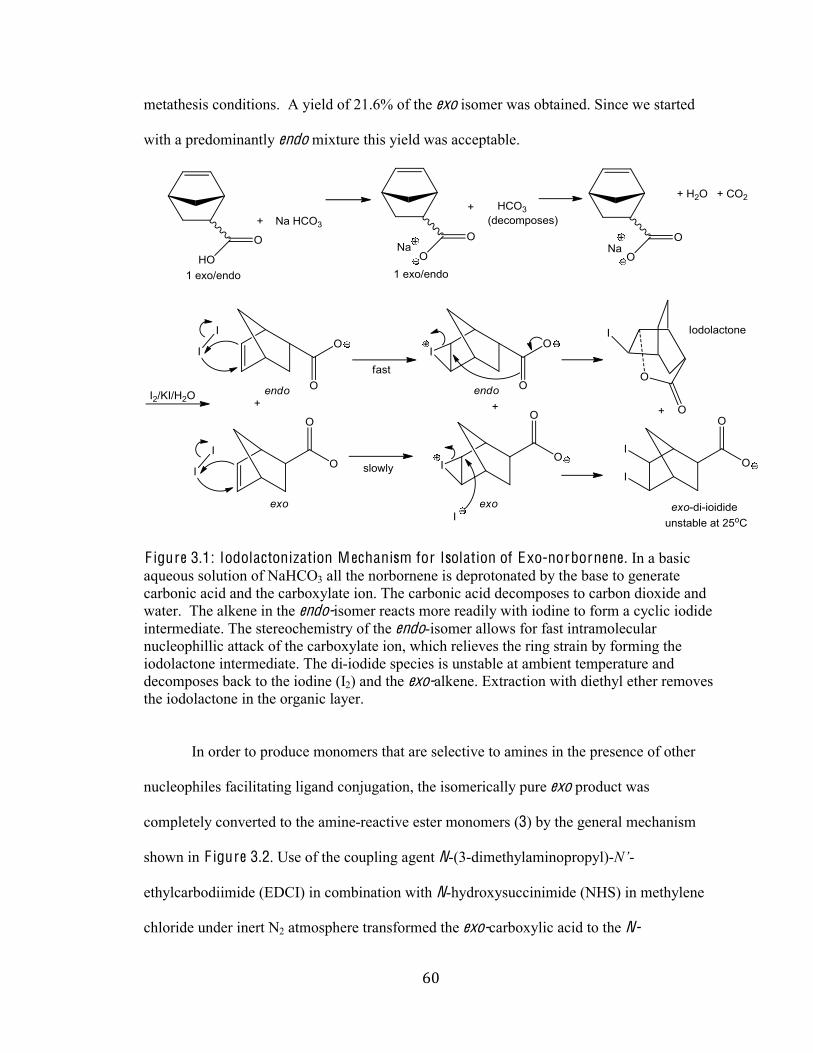
*.#
#
metathesis conditions. A yield of 21.6% of the exo isomer was obtained. Since we started
with a predominantly endo mixture this yield was acceptable.
F igure 3.1: Iodolactonization M echanism for Isolation of Exo-norbornene. In a basic
aqueous solution of NaHCO3 all the norbornene is deprotonated by the base to generate
carbonic acid and the carboxylate ion. The carbonic acid decomposes to carbon dioxide and
water. The alkene in the endo-isomer reacts more readily with iodine to form a cyclic iodide
intermediate. The stereochemistry of the endo-isomer allows for fast intramolecular
nucleophillic attack of the carboxylate ion, which relieves the ring strain by forming the
iodolactone intermediate. The di-iodide species is unstable at ambient temperature and
decomposes back to the iodine (I2) and the exo-alkene. Extraction with diethyl ether removes
the iodolactone in the organic layer.
In order to produce monomers that are selective to amines in the presence of other
nucleophiles facilitating ligand conjugation, the isomerically pure exo product was
completely converted to the amine-reactive ester monomers (3) by the general mechanism
shown in F igure 3.2. Use of the coupling agent N-(3-dimethylaminopropyl)-!"-
ethylcarbodiimide (EDCI) in combination with N-hydroxysuccinimide (NHS) in methylene
chloride under inert N2 atmosphere transformed the exo-carboxylic acid to the N-

*%#
#
succinimidyl ester substituted exo-norbornene monomer (3). EDCI activated the carboxylic
acid group of 2 forming an unstable O-acylisourea 2b that is more labile for the attack of the
NHS hydroxyl nucleophile. Acidification and extraction of the reaction mixture using citric
acid, allowed for the removal of the urea by-product 2c. The activated carboxylic acid ester
(3), was purified by flash column chromatography, to afforded an 85% yield and
subsequently analyzed by Proton Nuclear Magnetic Resonance (1H NMR) Spectroscopy.
Scheme 2: Subsequent Conversion of Exo-Norbornene to Succinimidyl Ester . The exo
isomer (2), is subsequently converted to the activated carboxylic acid ester, amine-reactive N-succinimidyl ester substituted exo-norbornene (3). Reagents and conditions: Isomerically
pure exo- norbornene acid (2) (1.22 g, 8.81 mmol, 1.0 eq), EDCI (2.53g, 13.2 mmol, 1.5 eq),
NHS (1.52 g, 13.2 mmol, 1.5 eq), CH2Cl2, 25o C, 24 h, 57.8%.
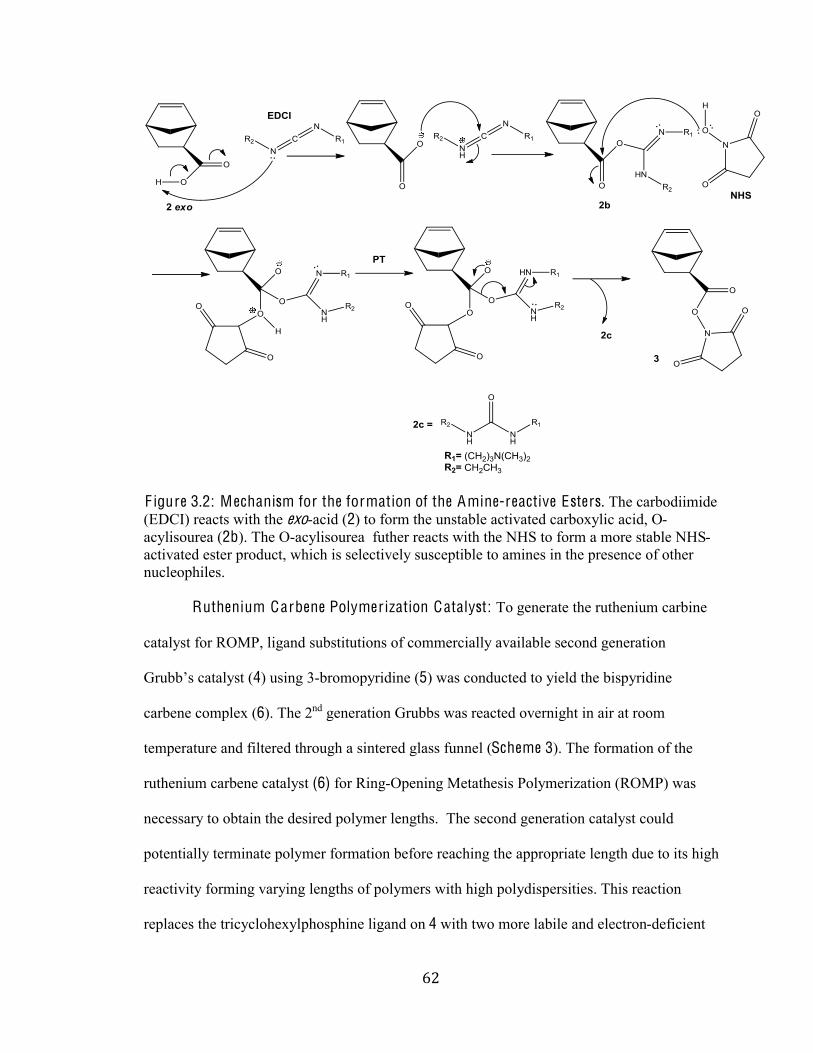
*&#
#
F igure 3.2: Mechanism for the formation of the Amine-reactive Esters. The carbodiimide
(EDCI) reacts with the exo-acid (2) to form the unstable activated carboxylic acid, O-
acylisourea (2b). The O-acylisourea futher reacts with the NHS to form a more stable NHS-
activated ester product, which is selectively susceptible to amines in the presence of other
nucleophiles.
Ruthenium Carbene Polymer ization Catalyst: To generate the ruthenium carbine
catalyst for ROMP, ligand substitutions of commercially available second generation
M)*;;"+%9-3-24+t (4) using 3-bromopyridine (5) was conducted to yield the bispyridine
carbene complex (6). The 2nd generation Grubbs was reacted overnight in air at room
temperature and filtered through a sintered glass funnel (Scheme 3). The formation of the
ruthenium carbene catalyst (6) for Ring-Opening Metathesis Polymerization (ROMP) was
necessary to obtain the desired polymer lengths. The second generation catalyst could
potentially terminate polymer formation before reaching the appropriate length due to its high
reactivity forming varying lengths of polymers with high polydispersities. This reaction
replaces the tricyclohexylphosphine ligand on 4 with two more labile and electron-deficient
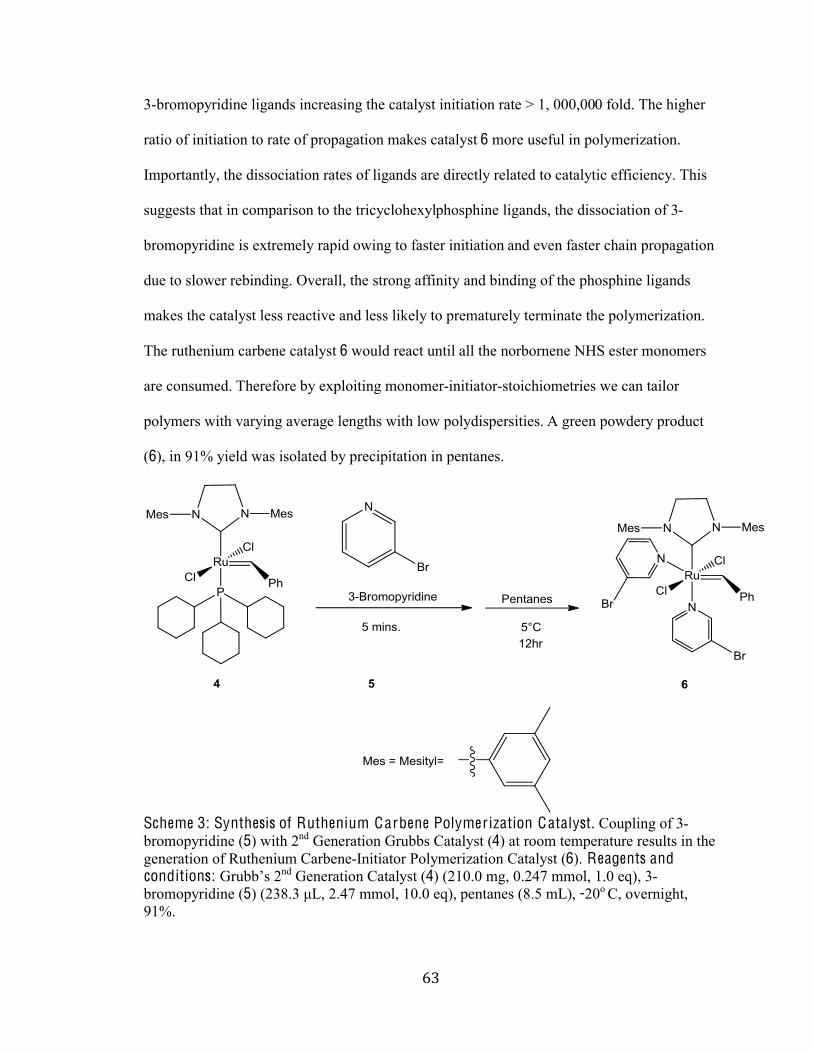
*'#
#
3-bromopyridine ligands increasing the catalyst initiation rate > 1, 000,000 fold. The higher
ratio of initiation to rate of propagation makes catalyst 6 more useful in polymerization.
Importantly, the dissociation rates of ligands are directly related to catalytic efficiency. This
suggests that in comparison to the tricyclohexylphosphine ligands, the dissociation of 3-
bromopyridine is extremely rapid owing to faster initiation and even faster chain propagation
due to slower rebinding. Overall, the strong affinity and binding of the phosphine ligands
makes the catalyst less reactive and less likely to prematurely terminate the polymerization.
The ruthenium carbene catalyst 6 would react until all the norbornene NHS ester monomers
are consumed. Therefore by exploiting monomer-initiator-stoichiometries we can tailor
polymers with varying average lengths with low polydispersities. A green powdery product
(6), in 91% yield was isolated by precipitation in pentanes.
Scheme 3: Synthesis of Ruthenium Carbene Polymer ization Catalyst. Coupling of 3-
bromopyridine (5) with 2nd Generation Grubbs Catalyst (4) at room temperature results in the
generation of Ruthenium Carbene-Initiator Polymerization Catalyst (6). Reagents and conditions: M)*;;"+%`nd Generation Catalyst (4) (210.0 mg, 0.247 mmol, 1.0 eq), 3-
bromopyridine (5) C`cPNc%hG,%`Nfe%@@02,%&UNU%1iB,%pentanes (8.5 mL), -20o C, overnight,
91%.

*(#
#
G eneral Succinimidyl Ester Substituted Polymeric Scaffold: The succinimidyl substituted
polymers was synthesized under inert nitrogen (N2) atmosphere by ring-opening-metathesis
polymerization using bispyridine-carbene catalyst (6) (Scheme 4). Control over average
polymer length was accomplished by variation of the monomer-to-initiator (M/I) catalyst
stoichiometric ratio. Ratios of 10:1 monomer: catalyst, 25:1, 50:1, and 100:1 were used to
obtain polymers of average lengths n~10, n~25, n~50 and n~100 (7a-d). A solution of NHS
ester monomer (3) dissolved in anhydrous (degassed) dichloromethane was cooled in a dry
ice/isopropanol bath to -`U%rQN%H61%8024@1)(\-3(0/%)1-93(0/%7-+%(/(3(-31:%*80/%-::(3(0/%0A%
solution of ruthenium catalyst (6) in degassed dichloromethane and termination after
complete consumption of the substituted norbornene (3) with ethyl vinyl ether, an electron
rich olefin. The excess ethyl vinyl ether undergoes metathesis with the living polymer chain
end carrying Grubbs catalyst to generate a metathesis-inactive Fischer carbene. After 12
hours, the polymer was precipitated into falcon tube from vortexing diethyl ether (Et2O).
After centrifuging and decanting the tubes, the precipitate was dried under high vacuum,
leaving a greenish solid for the 10-mer and grey solid for the 25-mer, 50-mer and 100-mer.
Polymerization yields ranged between 27 and 37%. The low percent yields were presumably
due to too much dicholoromethaneFa solvent which our polymers was soluble inFin the
vortexing tubes. Note that using more diethyl ether and/or more falcon tubes can optimize the
yields. The resulting polymers were characterized by 1H NMR spectroscopy to afford the
average length (Mn) values. The Mn values were determined by comparing the 1H-NMR
integration signals of the polymer alkene protons to that of the terminal phenyl protons
(Puffer 2007). The calculated Mn values more or less corresponded to the expected values
from the monomer initiator ratios, M:I =10:1, 25:1, 50:1, 100: 1.

*)#
#
Scheme 4: Synthesis of Succinimidyl Ester Substituted Polymer Scaffolds. Using
Ruthenium Carbene-Initiator Catalyst (6), succinimidyl ester polymers (7a-d) with the
average lengths n~10, n~25, n~50 and n~100 were derived from monomer units of N-succinimidyl ester substituted exo-norbornene (3). Reagents and conditions: i) 10-mer: Amine reactive ester (3) (200.0 mg, 0.8502 mmol 10 eq), Ruthenium Carbene-Initiator
Catalyst (6) (75.2 mg, 0.08502 mmol, 1.0 eq) -20oC, 30 mins, ethyl vinyl ether (3-5 drops),
24 hr, 27.4%. ii) 25-mer: Amine reactive ester (3) (200.0 mg, 0.8502 mmol 25 eq),
Ruthenium Carbene-Initiator Catalyst (6) (30.1 mg, 0.03401 mmol, 1.0 eq) -20oC, 30 mins,
ethyl vinyl ether (3-5 drops), 24 hr, 32 %. iii) 50-mer: Amine reactive ester (3) (200.0 mg,
0.8502 mmol 50 eq), Ruthenium Carbene-Initiator Catalyst (6) (15.0 mg, 0.0170 mmol, 1.0
eq), -20oC, 30 mins, ethyl vinyl ether (3-5 drops), 24 hr, 27.9%. iv) 100-mer: Amine reactive
ester (3) (200.0 mg, 0.8502 mmol 100 eq), Ruthenium Carbene-Initiator Catalyst (6) (7.52
mg, 0.008502 mmol, 1.0 eq), -20oC, 30 mins, ethyl vinyl ether (3-5 drops), 24 hr, 36.3%.
These polymers will be used in our EMSA assays.
Our next objective was to modify the succinimidyl substituted polymeric scaffolds
with our amine-bearing ligands of choice. To generate polymeric drugs to target the TAR
RNA, the amino acids arginine and agmatine were coupled to the polymers (Scheme 5). In
our experimental protocol, a 1.0-eq solution of NHS-polymer (7a-c) dissolved in dimethyl
sulfoxide (DMSO), 2.0 eq of Homo-S-Arginine was added, followed by 5.0 eq of N-
Methylmorpholine (NMM) at room temperature. N-Methylmorpholine is an organic base that
acts a coupling reagent to facilitate the formation of amide bond between the polymeric acyl
esters and the most reactive primary amine of the amino acids arginine and agmatine.
Overall, the synthesis of the amides in 8a-c and 9a-c proceeds via zwitterionic intermediate
that forms as a result of lone pairs on the amine attacking the carbonyl carbon, resolving its

**#
#
charge separation by collapse of the tetrahedral intermediate allowing elimination of the NHS
ester from the carbon center. The reaction mixture was then vortexed to facilitate dissolution
of all reagents. The reaction was then allowed to run for three days. Excess 3-amino-1, 2-
propanediol was used to convert any remaining N-hydroxysuccinimide (NHS) ester groups
into neutral functionality. The products were then purified and isolated using a PD-10
desalting size-exclusion column, eluted with Milli-Q water. PD-desalting columns separate
compounds by large differences in molecular weight. Globular or large molecular weight
compounds pass through column quickly, while compounds such as salts and small
molecules pass through slowly. Therefore, this technique provides an efficient way to
separate our large molecular weight polymeric products. Yields of the multivalent arginine
displays 8a-c ranged from 61-96%, while the yields from the agmatine displays ranged from
76-91% yield. As the polymer length increased the recovered yield decreased significantly.
The approximate weights of 8a-c polymers assuming 100% conjugation are 3000 Da, 8000
Da, and 16 000; while the weights of polymers 9a-c are 2800 Da, 7000 Da, and 14 000 Da.
These weights will be important when assessing the RNA-polymer complexes.

*+#
#
Scheme 5: Conjugation of Amines to Synthetic Polymer Scaffolds. Coupling of amine-
bearing guanidinums with the polymeric NHS ester. Quenching with excess 3-amino-1, 2
propanediol, terminates the reaction. Reagents and conditions: i) 8a-c: Respective
succinimidyl ester-substituted polymer 7a-d (5.3 mg, 0.0225 mmol, 1.0 eq), arginine (7.6 mg,
0.0437 mmol, 2.0 eq), N-Methylmorpholine (11.7 µL, 0.107 mmol, 5.0 eq) of, DMSO, 25 oC,
c:-4+,%cNJ%hG%c-amino propanediol, 61-96%. ii) 9a-c Respective succinimidyl ester-
substituted polymer 7a-d (5.3 mg, 0.0225 mmol, 1.0 eq), N-(4-Aminobutyl) Guanidine
(agmatine) (7.3 mg, 0.0561 mmol, 2.5eq) and N-Methylmorpholine (11.7 µL, 0.107 mmol,
5.0 eq),%>V?g,%c:-4+,%cNJ%hG%c-amino propanediol, 76-91%.
N-Norbornene-S-A rginine Monomer : In order, to make general comparisons
between the multivalent polymers and small molecules, conjugated monomers of arginine
were successfully synthesized (Scheme 6). The procedure for this synthesis was adapted and
modified from Choudhury. N-succinimidyl-ester substituted Norbornene (3) was added to a
solution of Homo-S-Arginine, dissolved in a 1:1 mixture of water-dioxane treated with
sodium bicarbonate and stirred at room temperature for 16 hr. Acidification, followed by
extraction with ethyl acetate isolated the crude product. The crude product was purified by
flash chromatography, which afforded a 96% yield of N-Norbornene-S-Arginine Monomer
(9). 1H NMR Spectroscopy was used to further assess the conversion as well as purity of the
product. All peaks corresponding to the alkene, amide and methylene protons were observed.
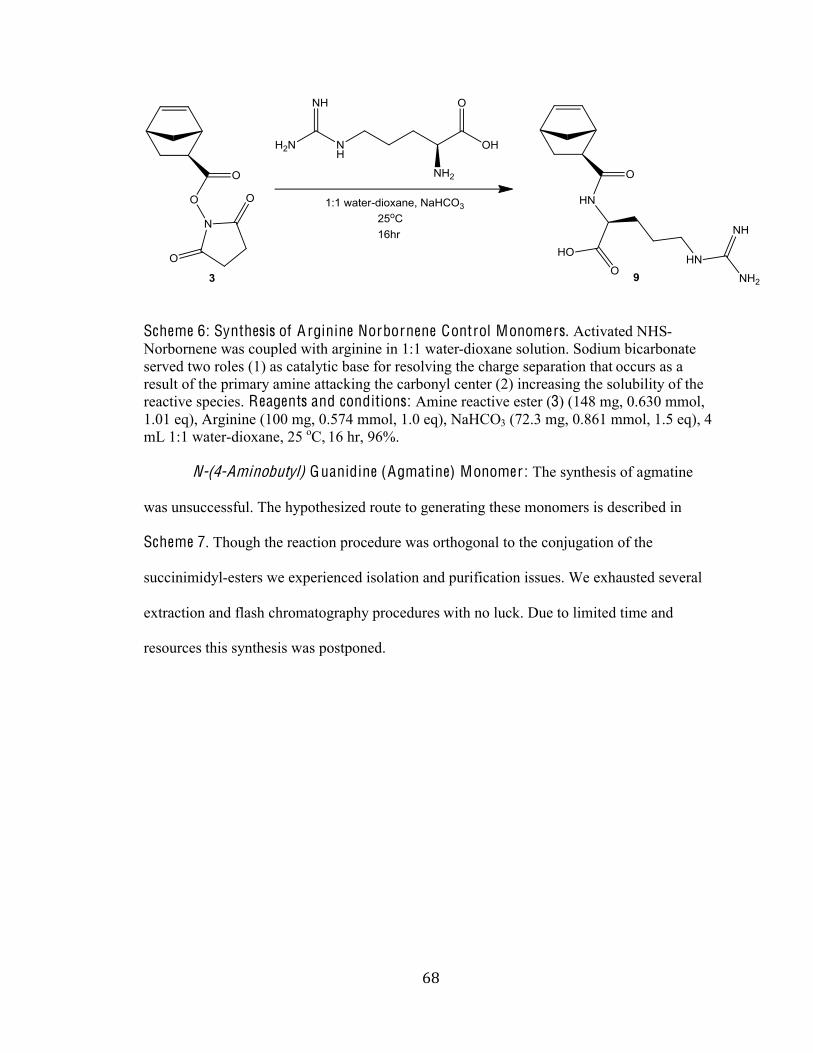
*,#
#
Scheme 6: Synthesis of A rginine Norbornene Control Monomers. Activated NHS-
Norbornene was coupled with arginine in 1:1 water-dioxane solution. Sodium bicarbonate
served two roles (1) as catalytic base for resolving the charge separation that occurs as a
result of the primary amine attacking the carbonyl center (2) increasing the solubility of the
reactive species. Reagents and conditions: Amine reactive ester (3) (148 mg, 0.630 mmol,
1.01 eq), Arginine (100 mg, 0.574 mmol, 1.0 eq), NaHCO3 (72.3 mg, 0.861 mmol, 1.5 eq), 4
mL 1:1 water-dioxane, 25 oC, 16 hr, 96%.
N-(4-Aminobutyl) Guanidine (Agmatine) Monomer : The synthesis of agmatine
was unsuccessful. The hypothesized route to generating these monomers is described in
Scheme 7. Though the reaction procedure was orthogonal to the conjugation of the
succinimidyl-esters we experienced isolation and purification issues. We exhausted several
extraction and flash chromatography procedures with no luck. Due to limited time and
resources this synthesis was postponed.

*-#
#
Scheme 7: G eneral route for the synthesis of Agmatine Monomers. Attempts to generate
the Agmatine monomers. Reagents and Conditions: Amine reactive ester (3) (20.6 mg,
0.0876 mmol, 1.097 1iB,%<E@-3(/1%C&PN`%@E,%UNUeOe%@@02,%&NU%1iB,%!VV%C`J%hG,%UN`&`%
@@02,%JNU%1iB,%fJU%hG%>V?g,%`f%6)- 3 days, 0%.
Summary: Altogether, we hypothesized that we would be able to compare the
arginine derivatives of arginine and agmatine to assess the role of the carboxylic acid in the
inhibition of Tat/TAR-RNA interactions. In addition, the variability in polymer length will
also allow us to identify whether the valency increases or decreases the affinity of the
potential RNA-binding drugs. To test this hypothesis, we will assay the RNA-binding
activities of our guanidinylated polymers using an EMSA-based approach. We expect that
since the polymers display guanidiniums an essential requisite for the electrostatic interaction
between the RNA and its endogenous cognate, we should observed some level alteration in
the electrophoretic mobility shift of TAR RNA especially at high concentrations of the
polymer.

+.#
#
Section I I : E lectrophoretic Mobility Shift Assays The identification of multivalent oligomers that bind TAR RNA with great affinity
and specificity could potentially serve as a potent therapeutic antiviral agent in the fight
against HIV/AIDS. To this end, our laboratory collaborated with Dr. Melissa Kosiniski-
Collins to develop a quantitative assay that allows us to analyze the effects of our ROMP-
derived synthetic polymers on TAR RNA. To investigate whether our synthetic polymeric
guanidinium compounds possess desired reactivity, we performed binding studies using the
Electrophoretic Mobility Shift Assay (EMSA) technique. To implicate TAR RNA folding
and activity, we first dissolved the lyophilized RNA in Tris/EDTA buffer, pH 7.5. In our
protocol, we incubated the wild type HIV-1 TAR RNA at biological cesQ, followed by
subsequent gel electrophoresis (4oC, 130V, 90 mins) to determine the optimal concentration
required for visualization with commercially acquired SYBR® Green Nucleic acid EMSA
stain. By titrating varying concentrations of RNA we observed the gel mobility shifts as
illustrated in F igure 3.3. Using the Typhoon 6410 Variable Mode Scanner, we concluded
that the SYBR® GREEN stain was sensitive to 43 ng of TAR RNA or more. We also
concluded that for future binding/inhibition experiments, TAR RNA concentrations between
200 ]300nM would be ideal.
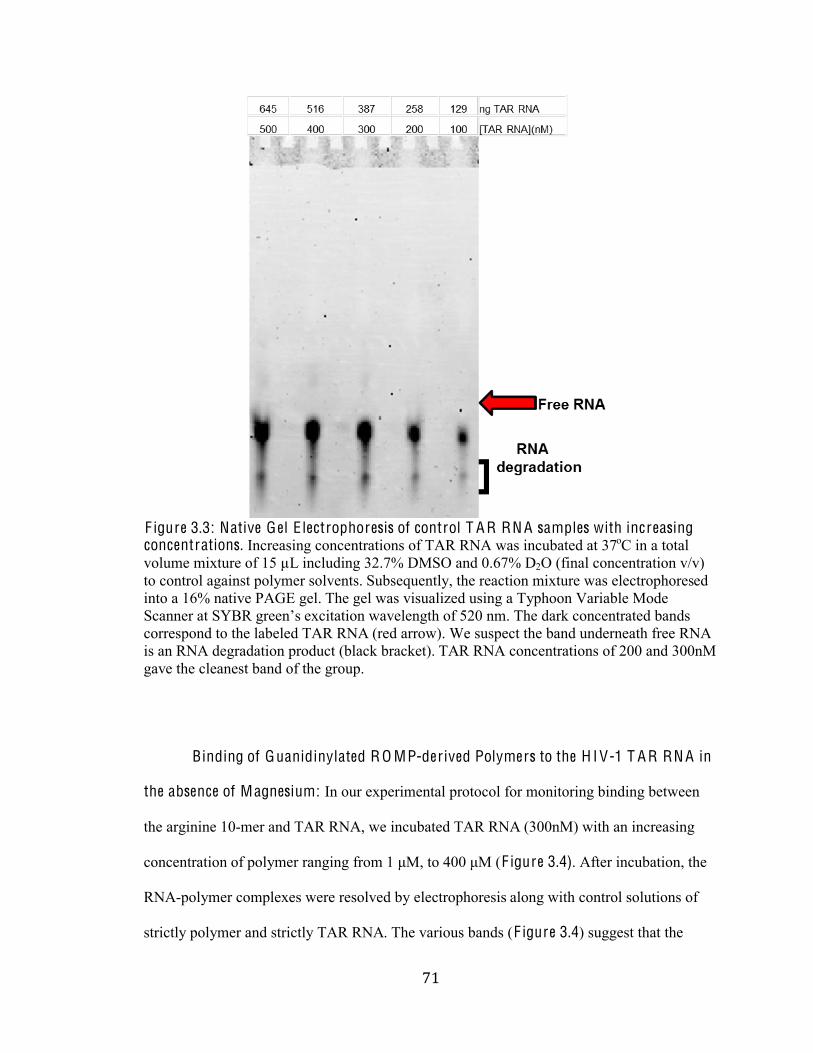
+%#
#
F igure 3.3: Native Gel E lectrophoresis of control T A R RN A samples with increasing concentrations. Increasing concentrations of TAR RNA was incubated at 37oC in a total
volume mixture of 15 µL including 32.7% DMSO and 0.67% D2O (final concentration v/v)
to control against polymer solvents. Subsequently, the reaction mixture was electrophoresed
into a 16% native PAGE gel. The gel was visualized using a Typhoon Variable Mode
Scanner at ?n$I%E)11/"+%excitation wavelength of 520 nm. The dark concentrated bands
correspond to the labeled TAR RNA (red arrow). We suspect the band underneath free RNA
is an RNA degradation product (black bracket). TAR RNA concentrations of 200 and 300nM
gave the cleanest band of the group.
Binding of Guanidinylated R O MP-derived Polymers to the H I V-1 T A R RN A in
the absence of Magnesium: In our experimental protocol for monitoring binding between
the arginine 10-mer and TAR RNA, we incubated TAR RNA (300nM) with an increasing
concentration of polymer ranging from &%hV,%30%fUU%hV%CF igure 3.4). After incubation, the
RNA-polymer complexes were resolved by electrophoresis along with control solutions of
strictly polymer and strictly TAR RNA. The various bands (F igure 3.4) suggest that the

+&#
#
arginine conjugated 10-mer binds the TAR RNA, decreasing its mobility through the gel.
Furthermore, the juxtaposition of the RNA-polymer complex band with the free RNA band
provided convincing evidence that arginine 10-mer is an RNA-binding molecule active in the
1-fUUhV%)-/E1N%The number of bands and their respective intensities are not sensitive to
small changes in polymer concentration, but large changes show an increase in the number of
visualized RNA-polymer complexes and a decrease in free RNA (Table 3.1). In the
concentration range of 1F10 µM, all the retarded band shifts were similar. In contrast, in the
range 200F400 µM we observed three or more high density oligomeric precipitates (F igure
3.4). The fluorescence in the retarded bands was quantified using a fluorescent Image Quant
software.
Table 3.1: Quantified A rginine 10-mer binding exper iment. This table corresponds to the
gel shifts in F igure 3.4. The gel shifts seen in the figure were categorized into free RNA and
complexed/bound RNA. The fluorescence is strictly a measurement of the SYBR stain on the
RNA, which was quantified using Imager Quant software. The values of Fluorescence are
reported in generic Absorbance Units (AU) as obtained from the Typhoon scanner. % bound
was calculated as complexed RNA AU divided by total absorbance times 100%.
10-mer Arginine:
d024@1)%ChVB
Free RNA
Fluorescence
Complex
Fluorescence % Bound
1 3449106 1704197 33.1
5 3510325 1685399 32.4
10 2761667 1260555 31.3
200 2423354 2090077 46.3
400 1708616 3653452 68.1
0 2793172 0 0
After confirming that the developed polymers bind TAR RNA, we inquired about the
binding mechanism of TAR RNA to the polymers. Initially, we hypothesized that since the
10-mer polymer is approximately the same length as the arginine rich domain in Tat, then the
multiple gel shift bands suggest that RNA-polymer interactions were non-specific. In
addition, such bindings could compel the RNA to fold into various configurations that could
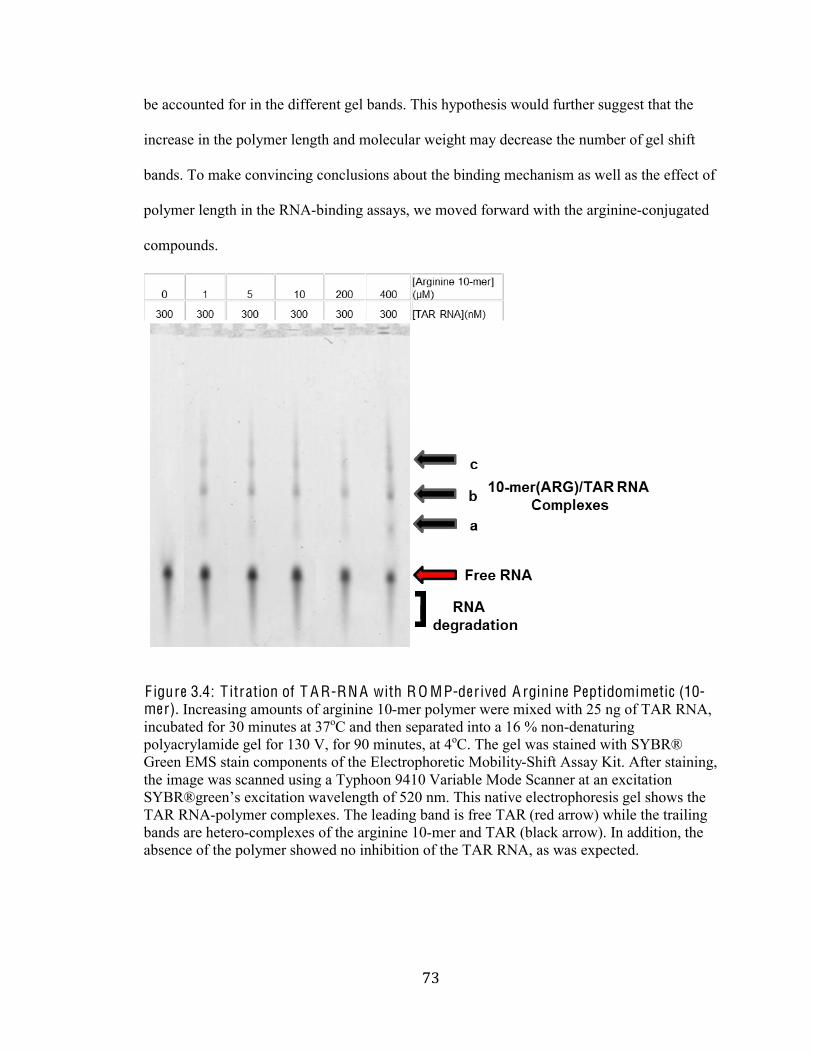
+'#
#
be accounted for in the different gel bands. This hypothesis would further suggest that the
increase in the polymer length and molecular weight may decrease the number of gel shift
bands. To make convincing conclusions about the binding mechanism as well as the effect of
polymer length in the RNA-binding assays, we moved forward with the arginine-conjugated
compounds.
F igure 3.4: T itration of T A R-RN A with R O MP-der ived A rginine Peptidomimetic (10-mer). Increasing amounts of arginine 10-mer polymer were mixed with 25 ng of TAR RNA,
incubated for 30 minutes at 37oC and then separated into a 16 % non-denaturing
polyacrylamide gel for 130 V, for 90 minutes, at 4oC. The gel was stained with SYBR®
Green EMS stain components of the Electrophoretic Mobility-Shift Assay Kit. After staining,
the image was scanned using a Typhoon 9410 Variable Mode Scanner at an excitation
?n$IoE)11/"+%1m9(3-3(0/%wavelength of 520 nm. This native electrophoresis gel shows the
TAR RNA-polymer complexes. The leading band is free TAR (red arrow) while the trailing
bands are hetero-complexes of the arginine 10-mer and TAR (black arrow). In addition, the
absence of the polymer showed no inhibition of the TAR RNA, as was expected.

+(#
#
In contrast to the arginine 10-mer, the bindings of arginine conjugated 25-mer and
50-mer to the wild type RNA in the 1F&U%hV%)-/ge resulted in only one shifted RNA band
(F igure 3.5-6). The results suggested that the 25-mer and 50-mer specifically recognize the
TAR RNA and retards the migration of the RNA. However, at considerably higher
concentrations (approximately 200!M), we begin to observe multiple retarded band shifts
suggesting less specific binding exist between the TAR RNA and the polymers. Given the
fact that the 25-mer and 50-mer are longer in length than the 10-mer, it is possible that
alternative polymer-RNA stoichiometries exist. The length would attribute multiple RNA
binding sites for a single polymer unit.
Table 3.2: Quantified A rginine 25-mer binding exper iment. This table corresponds to the
gel shifts in F igure 3.5. The gel shifts seen in the figure were categorized into free RNA and
complexed/bound RNA. The fluorescence is strictly a measurement of the SYBR stain on the
RNA, which was quantified using Imager Quant software. The values of Fluorescence are
reported in generic Absorbance Units (AU) as obtained from the Typhoon scanner. % bound
was calculated as complexed RNA AU divided by total absorbance times 100%.
25-mer Arginine:
d024@1)%ChVB
Free RNA
Fluorescence
Complex
Fluorescence % Bound
1 3448799 1202956 25.9
5 3705031 1002198 21.3
10 4131736 993494 19.4
200 2210197 1521484 40.8
0 4112013 0 0
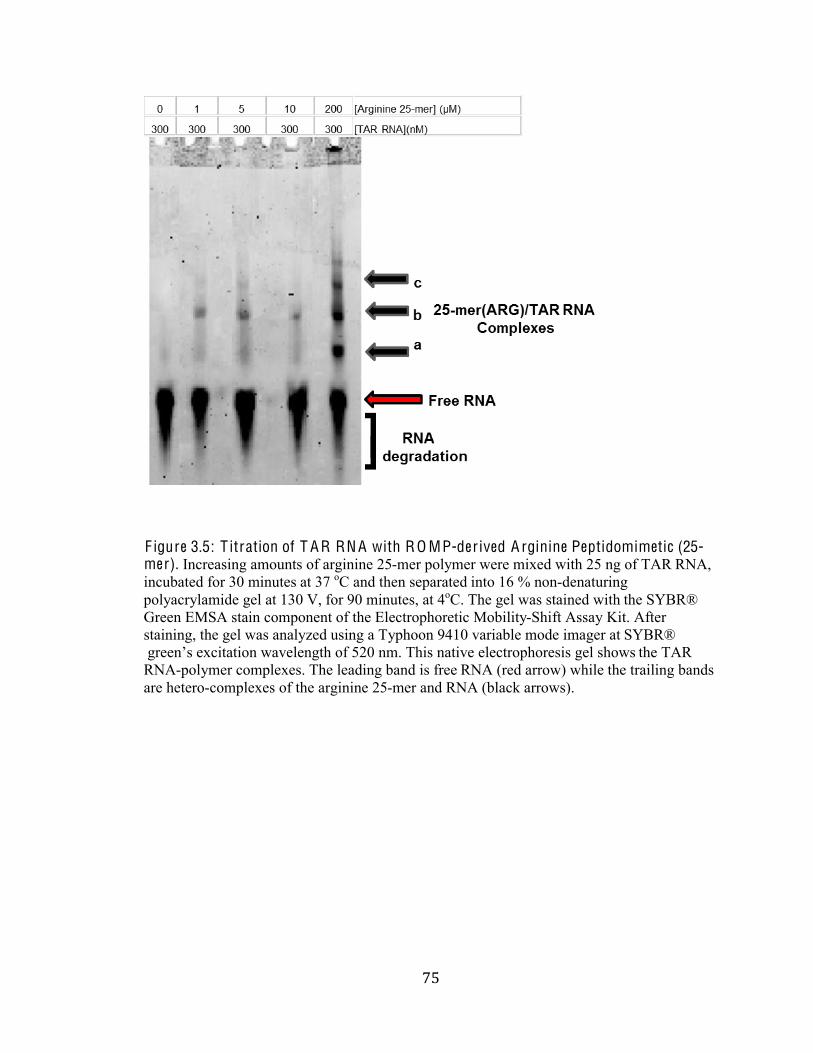
+)#
#
F igure 3.5: T itration of T A R RN A with R O MP-der ived A rginine Peptidomimetic (25-mer). Increasing amounts of arginine 25-mer polymer were mixed with 25 ng of TAR RNA,
incubated for 30 minutes at 37 oC and then separated into 16 % non-denaturing
polyacrylamide gel at 130 V, for 90 minutes, at 4oC. The gel was stained with the SYBR®
Green EMSA stain component of the Electrophoretic Mobility-Shift Assay Kit. After
staining, the gel was analyzed using a Typhoon 9410 variable mode imager at SYBR®
E)11/"+%excitation wavelength of 520 nm. This native electrophoresis gel shows the TAR
RNA-polymer complexes. The leading band is free RNA (red arrow) while the trailing bands
are hetero-complexes of the arginine 25-mer and RNA (black arrows).
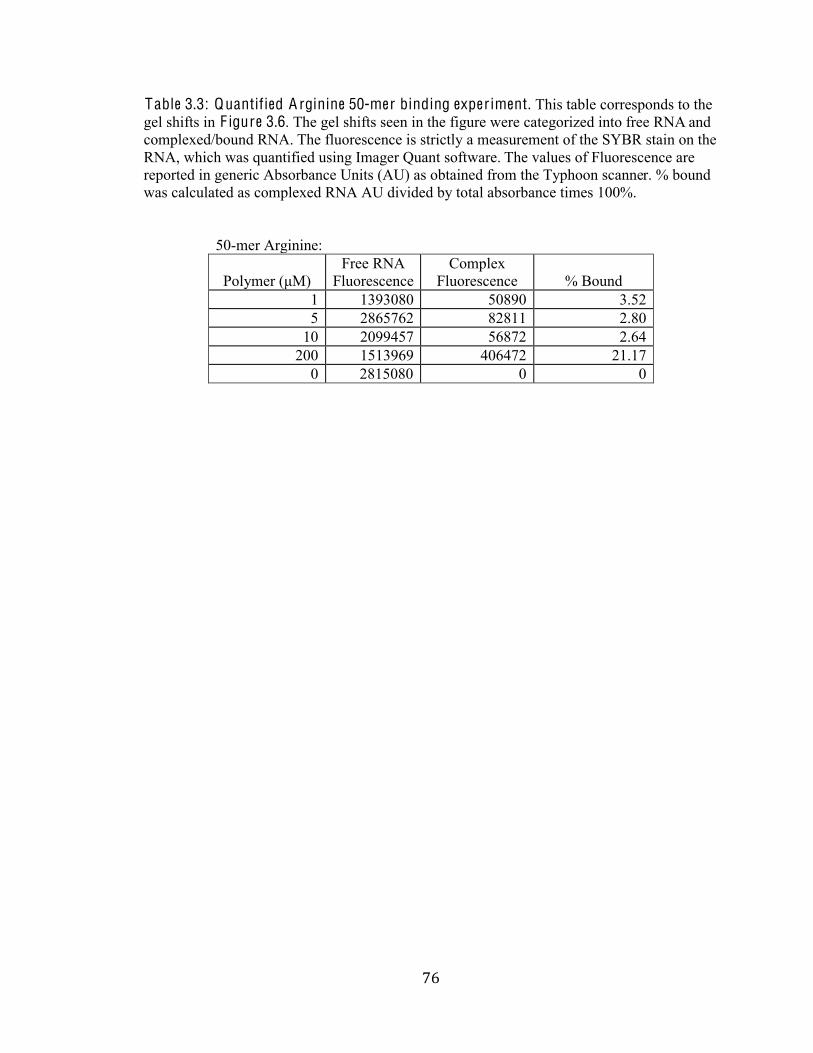
+*#
#
Table 3.3: Quantified A rginine 50-mer binding exper iment. This table corresponds to the
gel shifts in F igure 3.6. The gel shifts seen in the figure were categorized into free RNA and
complexed/bound RNA. The fluorescence is strictly a measurement of the SYBR stain on the
RNA, which was quantified using Imager Quant software. The values of Fluorescence are
reported in generic Absorbance Units (AU) as obtained from the Typhoon scanner. % bound
was calculated as complexed RNA AU divided by total absorbance times 100%.
50-mer Arginine:
Polymer (hVB
Free RNA
Fluorescence
Complex
Fluorescence % Bound
1 1393080 50890 3.52
5 2865762 82811 2.80
10 2099457 56872 2.64
200 1513969 406472 21.17
0 2815080 0 0
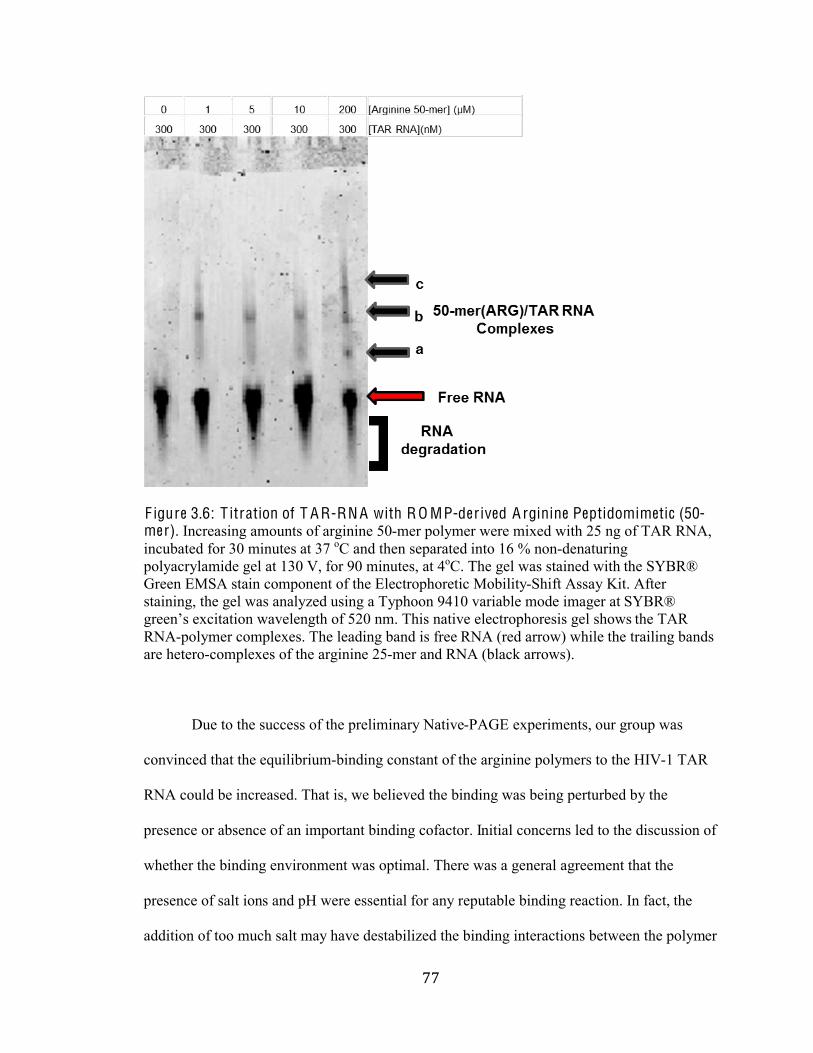
++#
#
F igure 3.6: T itration of T A R-RN A with R O MP-der ived A rginine Peptidomimetic (50-mer). Increasing amounts of arginine 50-mer polymer were mixed with 25 ng of TAR RNA,
incubated for 30 minutes at 37 oC and then separated into 16 % non-denaturing
polyacrylamide gel at 130 V, for 90 minutes, at 4oC. The gel was stained with the SYBR®
Green EMSA stain component of the Electrophoretic Mobility-Shift Assay Kit. After
staining, the gel was analyzed using a Typhoon 9410 variable mode imager at SYBR®
E)11/"+%excitation wavelength of 520 nm. This native electrophoresis gel shows the TAR
RNA-polymer complexes. The leading band is free RNA (red arrow) while the trailing bands
are hetero-complexes of the arginine 25-mer and RNA (black arrows).
Due to the success of the preliminary Native-PAGE experiments, our group was
convinced that the equilibrium-binding constant of the arginine polymers to the HIV-1 TAR
RNA could be increased. That is, we believed the binding was being perturbed by the
presence or absence of an important binding cofactor. Initial concerns led to the discussion of
whether the binding environment was optimal. There was a general agreement that the
presence of salt ions and pH were essential for any reputable binding reaction. In fact, the
addition of too much salt may have destabilized the binding interactions between the polymer

+,#
#
and RNA, decreasing the interactions actually observed on the gel. We concluded that the
initial 750 mM KCl binding buffer was concentrated enough to destabilize the binding
interactions. By lowering the salt concentration, we observed increased binding slightly, but
this enhancement was not significant enough to warrant the end to development of a better
binding reaction buffer. In fact, this lack of significant increase in binding led us to question
whether binding system promotes and maintains the native folding of the TAR RNA.
Incorrect folding may also have influenced the presence other bands in the gel.
To alleviate our concerns about the TAR RNA folding, as well as the hypothesized
multiple binding states of the various polymers, we decided to explore binding buffer systems
that contained both biological KCl and magnesium ion (Mg2+) concentrations. We then
studied the effects of introducing 50 mM KCl and 1 mM MgCl2 in the binding reaction and
same no evidence of misfolded or degraded RNA (Figure 3.7a). The effects of the Mg2+ ion
on the electrophoretic mobility of TAR RNA are shown in F igure 3.7b. Evidently, the
presence of the divalent Mg2+ cation in solution allowed for more sensitive binding between
TAR RNA and the polymers, with interactions being observed within polymer concentration
ranges of 1-10!M.
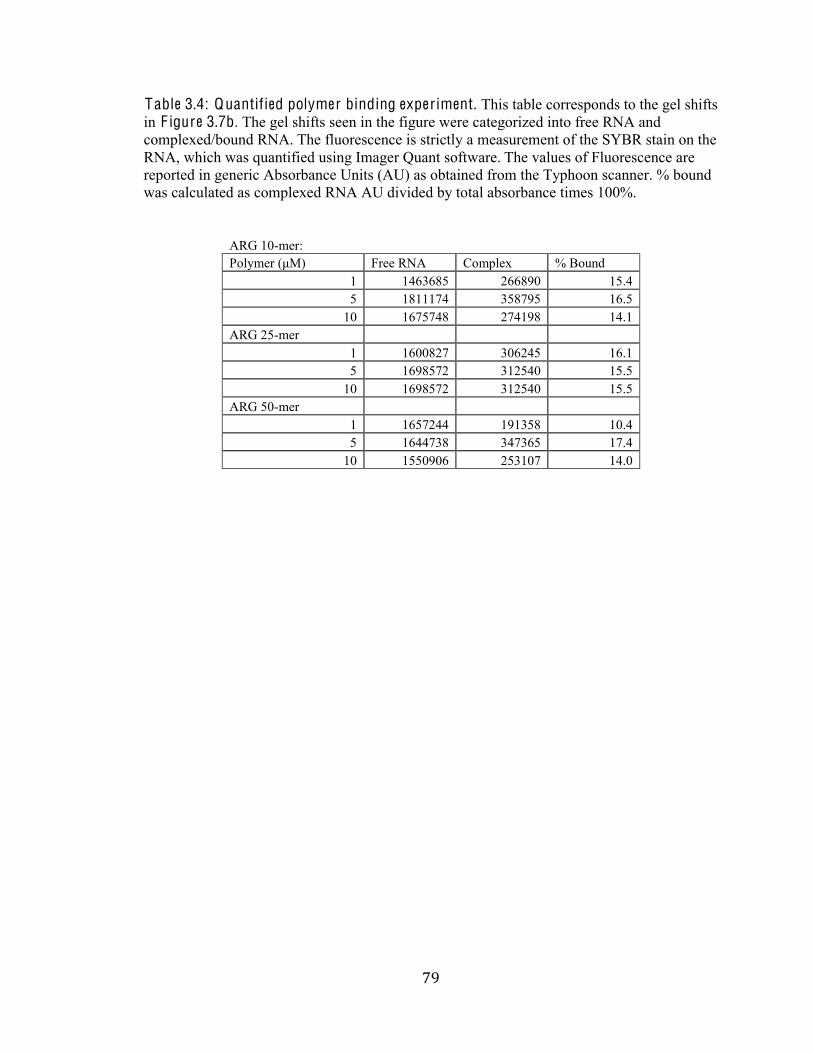
+-#
#
Table 3.4: Quantified polymer binding experiment. This table corresponds to the gel shifts
in F igure 3.7b. The gel shifts seen in the figure were categorized into free RNA and
complexed/bound RNA. The fluorescence is strictly a measurement of the SYBR stain on the
RNA, which was quantified using Imager Quant software. The values of Fluorescence are
reported in generic Absorbance Units (AU) as obtained from the Typhoon scanner. % bound
was calculated as complexed RNA AU divided by total absorbance times 100%.
ARG 10-mer:
d024@1)%ChVB Free RNA Complex % Bound
1 1463685 266890 15.4
5 1811174 358795 16.5
10 1675748 274198 14.1
ARG 25-mer
1 1600827 306245 16.1
5 1698572 312540 15.5
10 1698572 312540 15.5
ARG 50-mer
1 1657244 191358 10.4
5 1644738 347365 17.4
10 1550906 253107 14.0
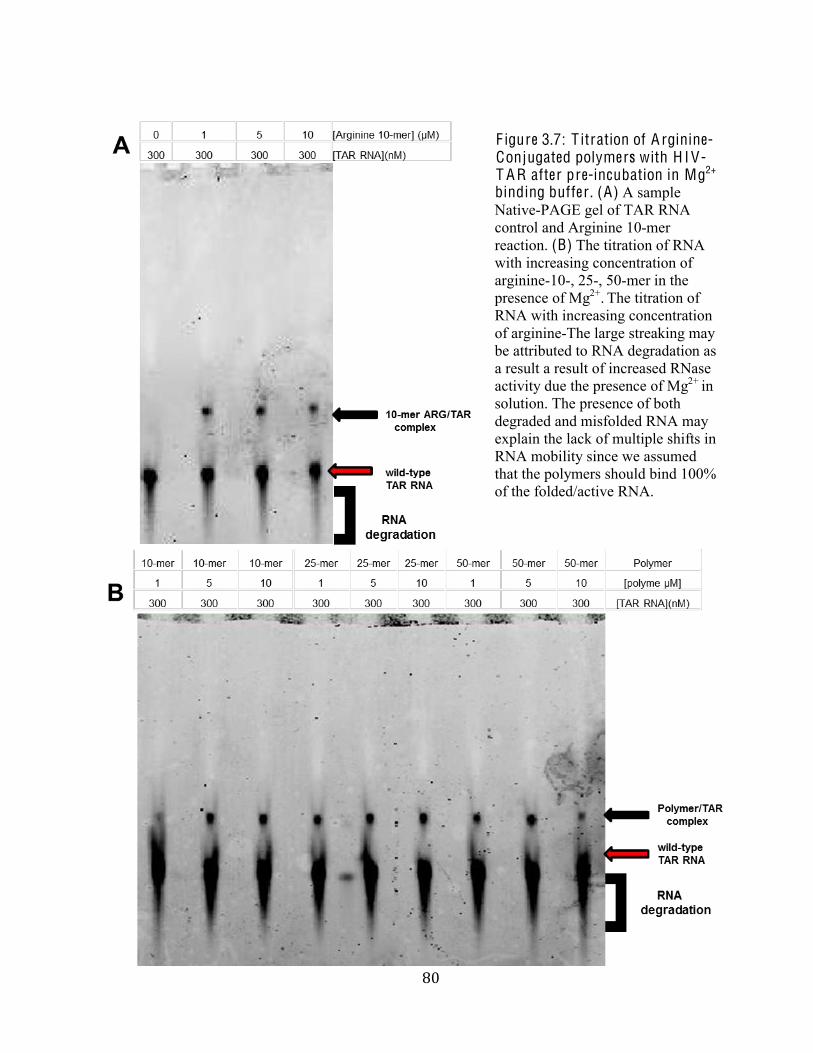
,.#
#
F igure 3.7: T itration of A rginine-Conjugated polymers with H I V-T A R after pre-incubation in Mg2+
binding buffer . (A) A sample
Native-PAGE gel of TAR RNA
control and Arginine 10-mer
reaction. (B) The titration of RNA
with increasing concentration of
arginine-10-, 25-, 50-mer in the
presence of Mg2+. The titration of
RNA with increasing concentration
of arginine-The large streaking may
be attributed to RNA degradation as
a result a result of increased RNase
activity due the presence of Mg2+ in
solution. The presence of both
degraded and misfolded RNA may
explain the lack of multiple shifts in
RNA mobility since we assumed
that the polymers should bind 100%
of the folded/active RNA.

,%#
#
To make further conclusions regarding whether our ROMP-derived polymers
possessed unique binding to the TAR RNA, we decided to evaluate the RNA-binding effects
of the free arginine and the arginine-conjugated norbornene monomer (9). The results of
these experiments under the optimized Mg2+ buffer conditions revealed multiple band shifts
for both the arginine-conjugated monomer and the free arginine (F igure 3.8). These results
suggest that the monomers may be affecting the mobility shift of the RNA in two ways: (1)
varying stoichiometric ratios of polymer and RNA, or (2) binding of the monomers forces the
TAR RNA to form a variety of tertiary structures. Both possibilities of large stoichiometries
or multiple configurations could cause the alteration of electrophoretic migration of TAR
RNA. In comparison, the separation of both the free arginine and the conjugated-arginine
norbornene monomer from free TAR RNA is orthogonal to those observed for the arginine
10-mer without the Mg2+. These results allow us to conclude that the arginine 10-mer
polymer binds more specifically to the HIV-1 TAR RNA than the monomers.
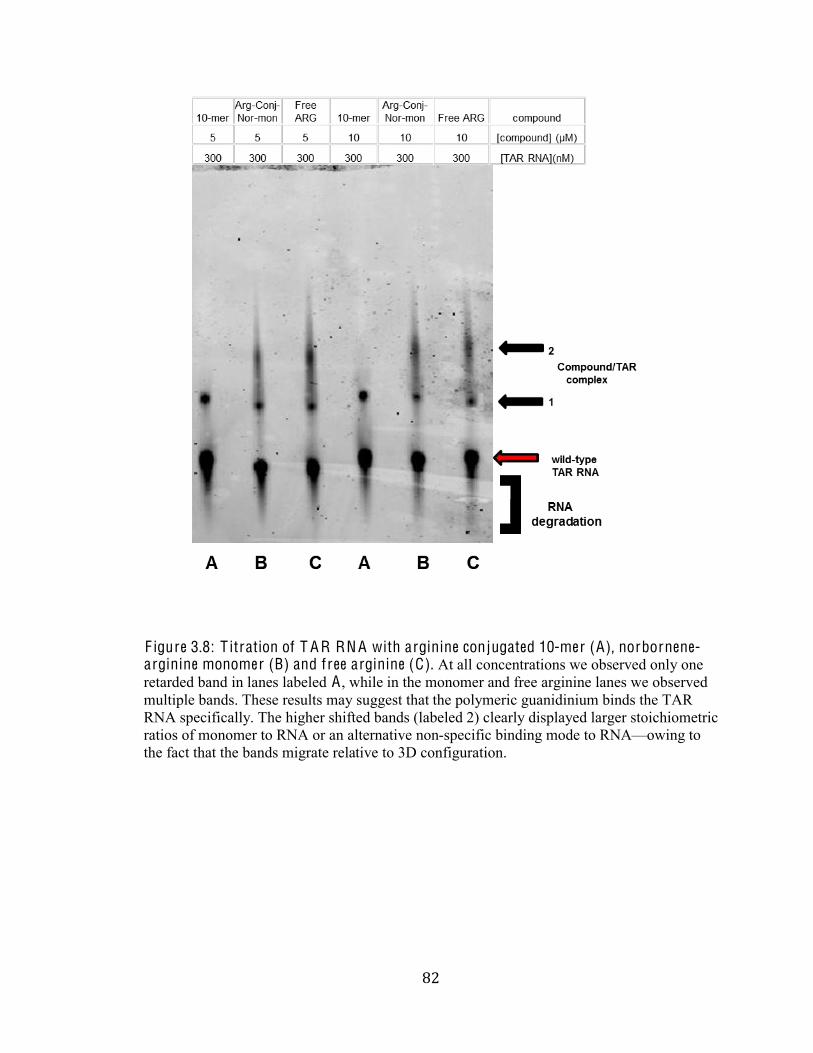
,&#
#
F igure 3.8: T itration of T A R RN A with arginine conjugated 10-mer (A), norbornene-arginine monomer (B) and free arginine (C). At all concentrations we observed only one
retarded band in lanes labeled A , while in the monomer and free arginine lanes we observed
multiple bands. These results may suggest that the polymeric guanidinium binds the TAR
RNA specifically. The higher shifted bands (labeled 2) clearly displayed larger stoichiometric
ratios of monomer to RNA or an alternative non-specific binding mode to RNAFowing to
the fact that the bands migrate relative to 3D configuration.

,'#
#
Discussion: Section I : Chemical Synthesis of Multivalent Guanidinium Inhibitors
Selective inhibition of the Tat/TAR interaction has long been considered an attractive
target for the development of potent antiviral agents. However, several caveats temper this
enthusiasm. First of all, it is difficult to inhibit viral protein-RNA interfaces with small
molecules. Secondly, scientists find it difficult to design RNA-binding molecules with well-
defined molecular recognition properties. Lastly, though numerous compounds have been
shown to bind TAR RNA none of these molecules warrant pharmaceutical development due
to non-specific binding, toxicity, poor biological activity (due to poor cellular uptake), or a
combination of these problems (Karn 1999; Yang 2005; Davidson 2009; Wang 2009). To
overcome some of these difficulties, we focused on a series of guanidinylated polymeric
peptide mimics of the Arginine Rich Motif (ARM) of the Tat protein, rationally developed
through structural considerations (Strong 1999; Karn 1999; Yang 2005; Davidson 2009). We
reasoned that the multivalency, and specificity of these polymers would show increased
selectivity for the TAR RNA compared to other reported polymeric peptidomimetics. In
addition, the large molecular weight of the polymer might allow for more potent and specific
interactions compared to small organic structures (Karn 1999; Yang 1999; Anthanassiou
2007; Davidson 2010; Tamilarasu 2001). Based on this rationale, we successfully
synthesized ROMP-derived polymeric guanidinium compounds that were characterized by
1H NMR Spectroscopy.
The synthesis of the reactive activated carboxylic ester monomer proceeded
efficiently (Scheme 1). Isomerically pure exo norbornene (2) was isolated from a

,(#
#
predominantly endo mixture of exo and endo 5-norbornene-2-carboxylic acid (1) using a
selective iodolactonization brew of I2 and KI in water to remove the excess endo isomer.
There was a 21.5% yield recovery, which is consistent with the amount of exo-stereoisomer
present in the starting reaction. The exo product in its entirety was converted to the amine-
reactive ester (3). The exo isomer was coupled with EDCI and NHS, followed by isolation
and purification using extraction and flash chromatography sequentially. A white solid was
recovered in 85% yield. It is important to note that the final yield and purity of the
Norbornene NHS ester 3 were optimized by selectively increasing the packing of the
stationary phase and/or varying the polarity of the eluent solvent during flash
chromatography. The high purity was reflected by the 1H NMR spectrum. All the expected
peaks were observed and distinguishable in the spectrum.
The synthesis of the bispyridine ruthenium carbene polymerization catalyst (6) from
the 2nd generation Grubbs catalyst (4) proceeded with great efficacy affording a colossal
yield of 91%. This yield was orthogonal to the published literature value of 89% (Love
2000). Owing to the fact that ring-opening metathesis polymerization can provide oligomers
of varying average lengths, the catalyst was employed in ROMP to yield the amine reactive
polymers 7a-d. By varying the ruthenium-carbene catalyst to monomer initiators ratios,
polymers 7a-d of varying lengths n~10, n~25, n~50 and n~100 were generated. All
polymerization reactions proceeded efficiently, consuming all the activated NHS ester
monomers. The acquired yields were 27%, 32%, 28% and 36% respectively. The resulting
solids were all grey in color except the 10-mer, which appeared greenish-grey. Though the
percent yields were low, there was more than enough grannular product acquired for the post-
polymerization ligand conjugations of the polymers with the guanidinium derivatives of
arginine and agmatine.

,)#
#
Post-synthetic modification of the amine-selective polymer scaffolds (7a-c) with
amine bearing guanidinums afforded products 8a-c and 9a-c. The polymers 7a-c were
dissolved in DMSO and coupled with arginine and agmatine to yield 8a-c and 9a-c
respectively. Initial issues with post modification occurred as a result of arginine and
agmatine insolubility in reaction solvent, DMSO. The literature reaction time for these post
polymerization modifications are on average 8 hrs (Kolonto 2009) but, due to the solubility
issue, the reaction time for our experiments was optimized to three days with sporadic
agitation by vortexing to increase collisions between the reactive species.
The yields of the conjugated polymers ranged from 61-96%. There was significant difference
in product yield based on length of polymer. In fact, recovery of the guanidinylated 100mer
was repeatedly unsuccessful. Successful conjugation was determined by 1H NMR
spectroscopy. Since the guanidinium signals are not distinguishable from the cyclopentane
polymer backbone, the percentile conversion was determined by integration of the methylene
signals of 3-amino-1-propanediol against the alkene signal of the polymer backbone (Kolonto
2009). We hypothesized that we would be able to compare the quandinium derivatives of
arginine and agmatine to assess the role of the carboxylic acid in the inhibition of Tat/TAR-
RNA interactions.

,*#
#
Section I I : Biological Assay
After successful synthesis, a biological assay that would give us both a qualitative
and quantitative assessment of the potency, selectivity and efficacy of our polymers in
binding HIV-1 TAR RNA needed to be developed. We hypothesized that the conjugated
ROMP-derived oligomers of arginine varying lengths can illuminate different biological
recognitions of the TAR RNA. Our understanding of the RNA-ligand systems suggested that
EMSA was the best method to afford us the preliminary and practical results we sought. In
this discussion, we will provide a chronological assessment and development of all the data
acquired from EMSA technique.
Preliminary Control Assays: Previous TAR-peptidomimetic binding assays
conducted by Mischiati et al., Davidson et al., and Bryson et al. revealed that the
concentration of TAR RNA required for observation of bound RNA complexes was
approximate range of 10F20 ng/nl (Mischiati 2001; Davidson 2009, Bryson 2012).
However, owing to the different preparatory schemes of the TAR RNA as well as labeling
dye, we decided to first conduct preliminary control assays with the TAR RNA. These
studies revealed that the minimum sensitivity of the commercially available SYBR ® Green
stain used in this study was approximately 43 ng of TAR RNA or more. Consequently, in
order to effectively assess inhibition we had to optimize the TAR RNA signal in the gel.
Optimization of the assay revealed that the optimal amount of TAR RNA was between 260
and 520ng for accurate quantitative analysis of the binding associations of TAR-
peptidomimetic polymer (F igure 3.3).
Binding of Arginine Conjugated Polymers to HIV-1 TAR RNA without Mg2+ : In the
first set of binding experiments, we analyzed our polymeric arginine compounds differing in

,+#
#
length. A single concentration of HIV-1 TAR RNA (300 nM) was incubated with these
compounds at concentrations ranging from &%hV to `UU%hV without Mg2+. After incubation,
RNA-polymer complexes were resolved by polyacrylamide gel electrophoresis. The initial
results of the argininge conjugated 10-mer polymers were promising (F igure 3.4). In both the
arginine conjugated 10-mer binding experiments, we saw that the mobility of a fraction of
TAR RNA migrated more slowly through the gel in comparison to the control. The altered
EMSA patterns revealed three retarded band shifts, which suggest that our compounds were
able to recognize and prevent the free migration of TAR RNA. From these initial binding
experiments of TAR RNA with arginine 10-mer, we envisaged that the bands represented
different oligomerization states, implying the possibility of three binding modes: (") multiple
RNA folds while bound, (ß) multiple molecules of TAR RNA per one polymerFa
consequence of zipping which may be dependent on polymer length, and (#) multiple
polymers per one RNA molecule. Please refer to the binding modes previously described for
the remainder of the discussion; they will be referred to as their letters ", ß, and #. All three
binding modes are possible given that in the microenvironment the binding mechanism of
RNA-polymer is unknown. Refer to F igure 4.1 for a detailed comparison of the proposed
binding modes.

,,#
#
F igure 4.1: Binding-modes of RN A-polymer . The possible modular binding mechanisms of
the ROMP-derived polymers to HIV-1 RNA as observed in the initial binding experiments of
arginine-10mer to TAR RNA. (A) Mechanism of binding mode ", indicating binding of
polymer to different RNA folding states. (B) Describes only binding mode ß, indicating
multiple polymers bound per RNA. (C) Description of binding mode #, indicating multiple
RNA per polymer. (D) Description the combination of binding modes ß and #. Kinetic
equations may be derived from these possible-binding mechanisms occurring between the
TAR RNA and the guanidinylated polymers. We hypothesized that the on and off rates Kon
and Koff of ligand binding may be severely dependent on the salt concentration, pH, and co-
factors such as Mg2+.
Given binding system ", we hypothesized that the binding between the ROMP-
derived 10-mer would not be specific or selective for biologically active TAR 3-nucleotide
bulge, a tertiary fold which is conserved over all strains of the HIV-1 virus (Aboul-ela 1996;
Karn 1999). This would suggest that the polymer did not simply attach to the TAR RNA
bulge but also to the negatively charged phosphates in the backbone of the misfolded TAR
RNA molecules. However, owing to these speculations and observations we reasoned that
with longer polymers (25-mer and 50-mer), we should observe multiple or fewer gel shift
bands, consequences of either binding mode " or # respectively. These two binding modes
can be modeled using the kinetic equations in F igure 4.1. These kinetic equations will

,-#
#
subsequently be used as a framework for our discussion regarding the bindings of 25-mer and
50-mer under conditions similar to the 10-mer.
Continued binding reactions with arginine-conjugated 25-mer and 50-mer provided
evidence suggesting that the polymer bindings are a result of at least binding mechanism".
We were unable to discern whether ß and # were also occurring. Furthermore, the polymers
have no tertiary structure and as a result we cannot determine whether the bindings of a 10-,
25-, or 50-mer travel differently through the gel. As we can see from comparing the gels in
F igures 3.4-6 (using the 200nM lane as the standard for comparison), all the gel shift bands
(labeled b) are all in the same position, reflecting that the bindings of either 10-, 25- or 50-
mer are the same and that the predominating factor in gel mobility is the folding state of the
RNA molecule while bound. To justify this reasoning, ongoing work seeks to stain Native-
PAGE gels containing the varying polymer lengths using Coomassie stain. This will
delineate further if the polymers have a tertiary conformation and whether they travel through
361%E12%-3%:(AA1)1/3%)-31+,%76(96%3614%+60*2:/"3
Major concerns about the binding modes compelled us to contemplate ways
to further parse out binding-modes", ß, and #. At first we strategized a test for binding mode
". This experiment would involve some form of crystallographic or Cryo Electron
Microscopy technique that we did not have the time or resources to test. In addition, in
absence of Mg2+, a few configuration of TAR RNA may exist. On the other hand, we were
very confident we could differentiate between the presence of binding mechanisms ß and #
using the following tests: (1) titration of a low concentration of RNA against a single
concentration of polymer and (2) simple mobility shift assay comparing properly folded TAR
RNA in the presence of excess 10-, 25-, and 50-mer polymer and all necessary biological
cofactors.

-.#
#
Binding of Arginine Conjugated Polymers in the presence of Mg2+ : Given our
previous results, we realized that a crucial factor in our experiments may have been
misfolding of the TAR RNA due to the lack of Magnesium ion in solution. In the biological
microenvironment, charged groups are essential for the binding interactions of the hetero-
complexes of protein-nucleic acids and vary due to the binding modes and charge
complementarities between the molecules. It is important to note that these interactions are
often electrostatically guided: molecules that are highly charged like the macromolecules
RNA and DNA are steered towards their interacting partners via the electric force that brings
the molecules together. That is, the interactions between native proteins and their
macromolecular cognate are specific and they can recognize each other among hundreds of
thousands of candidates (Wang 1996). Upon binding, there are induced ionization changes
due to the proton donating and accepting in ligand-macromolecule associations. However,
these proton donor/-991830)%@196-/(+@+%0)(E(/-31%A)0@%361%(/:('(:*-2%8D-"+%0A%3(3)-3-;21%
groupsF a consequence of pH and salt ion concentration in the microenvironment.
Therefore, different binding interactions can occur at different pH values. For example,
bovine-t-lactoglobulin complexation forms a dimer at low pH but forms a tetramer at high
pH. In this case Sakurai and coworkers showed that the addition of salts such as NaCl, KCl,
and NaClO4 stabilized the varying interaction states (Sakuri 2001). Consequently, changing
the salt concentration can cause the binding constant to change by several orders of
magnitude.
Acknowledging this phenomenon, we concluded that a crucial factor for the binding
reaction is the solution in which the binding reaction was performed. We theorized that
optimizing the ligand-protein electrostatic interactions required the use of an appropriate

-%#
#
buffer, pH, and salt concentration and to include any co-factors required for the interactions
involved. Due to the oligiomerization of the complexes observed from our assays, we
hypothesized that the presence of salts and other additives may reduce non-specific binding
of polymerFTAR RNA associations.
Initially, the addition of salts to the binding reaction provided inconclusive evidence
that buffer/salt crucially increased the on and off bindings of the polymeric guanidiniums to
TAR RNA. In fact, the presence of salt in our binding reaction resulted in little or no retarded
gel shift bands. This suggested that the salt destabilized the binding of the polymer to the
TAR by solvating it. Furthermore, cofactors crucial for the structural integrity and biological
activity and folding of RNA needed to be used. We discovered that we neglected to
incorporate the divalent cation magnesium (Mg2+) (Misra 1998, Misra 2002). Put simply,
rigorous studies with RNA showed that the stability of RNA tertiary structure is crucially
dependent on the concentration and presence of Mg2+ (Leipply 2010).
After pre-incubation of TAR RNA in 1 mM Mg2+ binding buffer and titration with
10-mer, 25-mer, and 50-mer arginine-conjugated polymers there was only one gel shift band
besides the free RNA observed on the gel. The absence of the multiple retarded bands
provides convincing evidence that the occupied RNA sustained multiple folding patterns
without the Mg2+ present in the binding reaction.
In contrast to the gel patterns generated in absence of Mg2+, we observed only one
retarded band for each of the polymer lengths (F igure 3.8). Despite the varying
concentrations of polymer and polymer length there was no difference in the percent RNAF
polymer bound (Table 3.4). At face value, these results suggest that the essential requisite for
binding of the three conjugated polymers is the presence of the guanidine moieties. This
observation reaffirms previous studies that the arginine rich motif of Tat recognizes HIV-1

-&#
#
TAR RNA. However, the single shifted band indicates one of two things: that the binding of
RNA to polymer is independent of the concentration and the valency of the polymer, or the
concentrations used in this experiment is significantly above the dissociation constant (KD).
It is expected that an increase in the concentration of the polymer with respect to
RNA should push the reaction forward, but this does not happen (F igure 4.2). As shown in
Table 3.4, all the tied up RNA-polymer complexes resulted in similarly resolved bands
despite the concentration of polymer. Theoretically, each polymer may have multiple binding
sites per RNA. At the concentrations worked with in this study, all the RNA should have
been bound due to polymer saturation. However, after three repetitions we saw that only
about 15-30% of the RNA was complexed with the polymers.
F igure 4.2: The equilibrium constant for the binding of ligand to single site. The KA is
also known as the association or affinity constant. [RNA*P] is the concentration of RNA-
polymer complex, [RNA] is the free concentration of TAR RNA and [P] is the free
concentration of polymer.
There is one other plausible conclusion that satisfies to this apparent independence of
concentration on the binding between the polymersFTAR RNA. That is, the retarded band
shift is indicative of only the correctly folded or active RNA such that we have already
saturated the RNA at concentrations 1µM of polymer. The fact that only a few percentage of
the RNA is in the viable active form is quite disappointing. Altogether, these results suggest
that the introduction of the magnesium into the binding buffer strongly enhances the
specificity and RNA-binding activities of the guanidinylated conjugates.
Comparing Monomers of Arginine to ROMP-derived 10-mer in presence of Mg2+: In
F igure 3.8 we saw observed only one retarded band shift for the Arginine-10mer, but for

-'#
#
arginine-conjugated norbornene and free arginine we observed two to three band shifts
suggesting that the monomers sequester RNA. Since the Arginine 10-mer does not sequester
the RNA as indicated by the one gel shift band, it can be further concluded that the polymer
binds to the RNA in a more specific manner as opposed to the monomers which binds the
RNA in multiple ways causing it fold differently. The different configurations of the bound
up RNA travel differently on the gel and results in the distinguished gel shift bands.

-(#
#
Conclusion:
In conclusion, the major finding in this work is the identification of RNA-binding
ROMP-derived guanidinylated polymers exhibiting structural affinity to the HIV-1 TAR
RNA. However, in other to improve the binding ratios of polymer to RNA we must conduct
further tests to optimize the binding reaction buffer. A reputable binding buffer solution will
provide us with the means to calculate binding constants such as KA. In addition, we may be
able to conduct subsequent tests to evaluate the selectivity and specificity of the
guanidinylated polymers. In addition, it is expected that TAR RNA binding molecules
potentially inhibits the association of Tat/TAR. To verify this expectation, ongoing work
seeks to study the effects of the ROMP-derived polymers on the Tat/TAR interactions using
EMSA.

-)#
#
Future Directions:
Targeting the Tat-peptide represents a parallel strategy to inhibit the association of
Tat/TAR-RNA. We plan to functionalize our various length polymer scaffolds to display
sulfates or carboxylates. We hypothesize that these anions will electrostatically interact with
the basic ARM of Tat thus preventing the association of Tat to TAR-RNA 3 base bulge.
Sulfate amines such as sulfanilic acid and 5-amino-1-pentane sulfonic acid are ideal because
they have both anionic and amine functionality optimal for exploration. The difference in
carbon lengths will allow for the exploration of proximity between the polymer scaffold and
sulfonic groups. A similar assessment may also be conducted between the carboxylic acid
amines of 4-aminomethyl benzoic acid and caproic acid.

-*#
#
Bibliography:
Anand, K.; Schulte, A.; Vogel-$-96@-4),%DNT%?961AA\1p,%DNT%M141),%VN%.?3)*93*)-2%(/+(E63+%
into the Cyclin T1]Tat]H<I%I!<%3)-/+9)(83(0/%-93('-3(0/%90@821m%A)0@%a=<LN5%
Nat. Struct. Mol. Biol. 2008, 15(12), 1287-1292.
Aboul-ela, F.; Karn, J.; Va)-/(,%MN%.?3)*93*)1%0A%K=L-1 TAR RNA in the absence of ligands
)1'1-2+%-%/0'12%90/A(E*)-3(0/%0A%361%3)(/*92103(:1%;*2E1N5%Nucl. Acids Res. 1996, 24,
3974]3981.
Aboul-12-,%qN%.?3)-31E(1+%A0)%361%:1+(E/%0A%I!<-;(/:(/E%+@-22%@0219*21+N5%Future Med. Chem. 2010, 2(1), 93-119.
Archin, N. M.; Liberty, A. L.; Kashuba, A. D.; Choudhary, S. K.; Kuruc, J. D.; Crooks, A.
M.; Parker, D. C.; Anderson, E. M.; Kearney, M. F.; Strain, M. C.; Richman, D. D.;
Hudgens, M. G.; Bosch, R. J.; Coffin, J. M.; Eron, J. J.; Hazuda, D. J.; Margolis, D.
VN%.<:@(/(+3)-3(0/%0A%'0)(/0+3-3%:(+)*83+%K=L-1 latency in patients on antiretroviral
361)-84N5%Nature 2012, 487, 482-489.
Amendt, B. A.; Hesslein, D.; Chang, L. J.; Stoltzfus, C. M. .d)1+1/91 of negative and
positive cis-acting RNA splicing elements within and flanking the first Tat coding
exon of the Human Immunodeficiency Virus Type-&N5 Mol Cell Biol. 1994, 14,
3960]3970.
$1)3-E/02(0,%<NT%u0):-/,%VN%INT%$-)9-)20,%uNT%d-)p(/,%!N%.R0)2:%K1-236%g)E-/(\-3(0/5%H61%
HIV Drug Resistance Report -`U&`N5%WHO, 2012; pp 1-84.
Bilodeau, P. S.; Domsic, J. K.; Stoltzfus,%VN%QN%.Splicing Regulatory Elements
within tat Exon 2 of Human Immunodeficiency Virus Type 1 (HIV-1) are
characteristic of Group M but not Group O HIV-&%?3)-(/+N5 J. Virol. 1999, 73(12), 9764]9772.
$)-99(,%GNT%q-29(-/(,%QNT%G122(,%$N%.?4/3613(9%8183(:1+%(/%361%A0)@%0A%:1/:)(@1)+%;190@1%
)1+(+3-/3%30%8)031-+1%-93('(34N5%J. Biol. Chem. 2003, 278, 46590]46595.
Bryson, D. I.; Zhang, W.; Ray, W. K.; Santos, W. L. .Screening of a branched peptide library
with HIV-1 TAR RNAN5%Mol. BioSyst. 2009, 5, 1070-1073.
$)4+0/,%>N%=NT%v6-/E,%RNT%V9G1/:0/,%dN%VNT%I1(/1p1,%HN%VNT%?-/30+,%RN%GN%.H07-):%
Targeting RNA Structure: Branched Peptides as Cell-Permeable Ligands to TAR
I!<N5%ACS. Chem. Biol. 2012, 7(1), 210-217.

-+#
#
Bugatti, A.; U)(/-3(,%QNT%I-'122(,%QNT%>1%Q21)9i,%aNT%G(1p1/+,%?NT%I*+/-3(,%VN%.K18-)(/-
Mimicking Sulfonic Acid Polymers as Multitarget Inhibitors of Human
=@@*/0:1A(9(1/94%L()*+%H481%&%H-3%-/:%E8&`U%d)031(/+N5%Antimicrobial Agents and Chemotherapy 2007, 51(7), 2337-2345.
Q-0,%KNT%H-@(2-)-+*,%!NT%I-/-,%HN%VN%.g)(1/3-3(0/%-/:%<AA(/(34%0A%K=L-1 Tat Fragments in
H-3wH<I%Q0@821m%>131)@(/1:%;4%q2*0)1+91/91%I1+0/-/91%a/1)E4%H)-/+A1)N5%
Bioconjugate Chem. 2006, 17, 352-58.
Chevallet, M.; Luche, S.; Rabilloud, HN%.Silver staining of proteins in polyacrylamide gels.5%
Nat. Protoc. 2006, 1(4), 1852]1858.
Choudhury, A. K.; Golovine, S. YNT%>1:p0'-,%GN%VNT%K1963,%?N%VN%.?4/361+(+%0A%d)031(/+%
Q0/3-(/(/E%V0:(A(1:%<)E(/(/1%I1+(:*1+N5%Biochemistry 2007, 46, 4066-4076
Q)071,%!N%nNT%M0:A)14,%>N%=NT%$-m31),%<N%MN%.!-3*)1%p(221)%H%9122+%-)1%3-)E13+%A0)%K*@-/%
=@@*/0:1A(9(1/94%'()*+%(/A193(0/N5%Immunology 2003, 108, 1-2.
Cullen, B. R. Transactivation of human immunodeficiency virus occurs via a bimodal
mechanism. Cell 1986, 46, 973-982.
Davidson, A.; Patora-Komisarska, K.; Robinson, J. A; Varani, GN%.a++1/3(-2%+3)*93*)-2%
requirements for specific recognition of HIV TAR RNA by peptide mimetics of Tat
8)031(/N5 Nucleic Acid Res. 2011, 39(1), 248-256.
>176*)+3,%?NT%M12;-):,%KN%<NT%q(/1,%?N%VN%.!1*)08-360E1/1+(+%0A%<=>?N5%Mol. Med. 1996, 2,
16-23.
Dingwall, C.; Ernberg, I.; Gait, M. J.; Green, S. M.; Heaphy, S.; Karn, J.; Lowe, A. D.;
Singh, M.; Skinner, M. A.; Valerio, R. (Human immunodeficiency virus 1 Tat protein
binds transactivation-responsive region (TAR) RNA in vitro. Proc.Natl Acad. Sci. USA 1989, 86, 6925-6929.
Ellis, J. S.; Thompson, M. .Q0/A0)@-3(0/-2%+3-31+%0A%/*921(9%-9(:]peptide complexes
@0/(30)1:%;4%-90*+3(9%7-'1%8)08-E-3(0/%-/:%@0219*2-)%:4/-@(9+%+(@*2-3(0/N5%Chem. Sci. 2011, 2, 237-255.
q-29(-/(,%QNT%G0\\(,%GNT%d(/(,%<N%.V0219*2-)%;-+(+%0A%;)-/961:%8183(:1+%)1+(+3-/91%30%1/\4@e
8)031024+(+N5%Chem. Biol. Drug. Des. 2007, 69, 216]221.
Fiala, M.; Popik, W.; Qiao, J. H.; Lossinsky, A. S.; Alce, T.; Tran, K.; Yang, W.; Roos, K.
dNT%%<)360+,%uN%.K=L-1 induces cardiomyopathy by cardiomyocyte invasion and gp120, Tat, and cytokine apoptoti9%+(E/-2(/EN5%Cardiovasc. Toxicol. 2004, 4, 97-107.
Fleuridor, R.; Wilson, B.; Hou, R.; Landay, A.; Kessler, H.; Al-Harth(,%GN%.CD1d-restricted
natural killer T cells are potent targets for human immunodeficiency virus infection.5 Immunology 2003, 108 (1), 3-9.

-,#
#
Fricker, S.P.; Anastassov, V.; Cox, J.; Darkes, M.C.; Grujic, O.; Idzan, S.R.; Labrecque, J.;
Lau, G.; Mosi, R.M.; Nelson, K.L.; Qin, L.; Santucci, Z.; Wong, R.S.Y.
.Q6-)-931)(\-3(0/%0A%361%@0219*2-)%86-)@-9020E4%0A%<V>c&UU^%<%+819(A(9%
antagonist of the G-8)031(/%90*821:%Q61@0p(/1%)191830),%QlQIfN5%Biochemical Pharmacology 2006, 72(5), 588-596.
Garber, M. E.; Wei, P.; Kewal-Ramani, V.; Mayall, T. P.; Herrmann, C. H.; Rice, A. P.;
Littman, D. R.; Jones, K. A. .The interaction between HIV-1 Tat and human cyclin
T1 requires zinc and a critical cysteine residue that is not conserved in the murine
CycT1 proteinN5%Genes Dev. 1998a, 12, 3512-3527
M-);1),%VN%aNT%R1(,%dNT%u0/1+,%DN%<N%.HIV-1 Tat Interacts with Cyclin T1 to Direct the P-
Haq;%QH>%D(/-+1%Q0@821m%30%H<I%I!<N5 Cold Spring Harb. Symp. Quant. Biol. 1998b, 63, 371-380.
M1+37(9p(,%uN%aN,%G-*)-%GN%DN%.=/31)-receptor Communication through Arrays of Bacterial
Q61@0)191830)+N5%Nature 2002, 415, 81-84.
Harrich, D.; Mavankal, G.; Mette-Snider, A.; Gaynor, R. B. .Human immunodeficiency virus
type 1 TAR element revertant viruses define RNA structures required for efficient
viral gene expression and replication.5 J. Virol. 1995, 69(8), 4906]4913.
Harrich, D.; Ulich, C.; Gaynor, R. B. .A critical role for the TAR element in promoting
efficient human immunodeficiency virus type 1 reverse transcription.5 J. Virol. 1996,
70, 4017-4127.
K*-/E,%KN%RNT%R-/E,%DN%HN%.?3)*93*)-2%96-)-931)(\-3(0/%0A%361%@13-2%;(/:(/E%+(31%(/%361%
cysteine-rich region of HIV-&H-3%8)031(/N5%Biochem. Biophys. Res. Commun. 1996,
227, 615-621.
Jeang, K. T.; Chun, R.; Lin, N. H.; Gatignol, A.; Glabe, C. G.; Fan, H. .=/%'(3)0%-/:%(/%'('0%
binding of human immunod1x9(1/94%'()*+%3481%&%H-3%8)031(/%-/:%?8&%3)-/+9)(83(0/%
A-930)N5%J. Virol. 1993, 67, 6224]6233.
u1-/E,%DN%HN%.K=L%-/:%Q122*2-)%q-930)+N5%%Molecular Virology Section 1996, 0, 1-13.
Jeang, K. T.; Xiao, H.; and Rich, E.A. (1999). .Multifaceted activities of the HIV-1
transactivator of transcription Tat.5 J. Biol. Chem. 1999, 274, 28837]28840.
Karn, J. "Tackling Tat." J. Mol. Biol. 1999, 293(2), 235-254.
Khalil,%<N%VNT%I(@,%uN%GN%.RNA-Protein Interactions (/%K*@-/%K1-236%-/:%>(+1-+1N5 Semin. Cell. Dev. Biol. 2011, 22(4), 359]365.
Kolonto, E. M.; Pontrello, J. K.; Mangold, S. L.; -/:%D(1++2(/E,%GNGN%.%M1/1)-2%?4/3613(9%
I0*31%30%Q122%d1)@1-;21%$209p%Q08024@1)+N5%J. Am. Chem. Soc. 2009, 131, 7327-
7333.

--#
#
D)1;+,%<NT%G*:7(E,%LNT%$0:1/,%gNT%V(96-12%RN%MN%.Targeting the HIV Trans-Activation
Responsive Region]Approaches Towards RNA-Binding Drugs.5Chem. Bio. Chem
2003, 4, 972-978.
Leipply, D. Draper, D. E. .The dependence of RNA tertiary structure stability on Mg2+
concentration: interpretation of the Hill equation and 901AA(9(1/3N5%Biochemistry 2010,
49(9), 1843]1853.
G(30'96(9p,%<NT%G-8(:03,%<NT%a(+1/+31(/,%VNT%D-2(/p0'(96,%<NT%$0)p0',%MN%.!10@49(/%$-
Arginine Conjugate, a Novel HIV-1 Tat Antagonist: Synthesis and Anti- HIV
<93('(3(1+N5 Biochemistry 2001, 40, 15612-15623.
Love, uN%<NT%V0)E-/%uNdNT%H)/p-,HNVNT%M)*;;+,%INKN%%.<%d)-93(9-2%-/:%K(E624%<93('1%
Ruthenium-$-+1:%Q-3-24+3%36-3%aAA193+%361%Q)0++%V13-361+(+%0A%<9)420/(3)(21N5%
Angew. Chem. Int. Ed. 2002, 41(21), 4035-4037.
!00/-/,%>N%-/:%<2;(/(,%<N%.q)0@%361%0*3+(:1%(/^%am3)-9122*2-)%-93('(3(1+%0A%K=L%H-3N5Adv. Pharmocol. 2000, 48, 229-250.
Mishra, Subrata H. "Structure and Energetics of RNA - Protein Interactions for HIV RREIIB
Targeting Zinc Finger Proteins." (2008). Chemistry Dissertations. Paper 22.
http://digitalarchive.gsu.edu/chemistry_diss/22/
V(+)-,%LN%DNT%>)-81),%>N%aN%.g/%361%I021%0A%V-E/1+(*@%=0/+%(/%I!<%?3-;(2(34N5%Biopolymers (Nucleic Acid Sciences) 1998, 48, 113]135.
Misra, V. K.; Draper, D. E. .The linkage between magnesium binding and RNA folding.5%J. Mol. Biol. 2002, 317(4), 507-521.
V(4-*96(,%DNT%D(@,%nNT%G-3(/0'(9,%gNT%V0)0\0',%LNT%V12(p4-/,%MN%$N%.K=L%1/31)+%9122+%'(-%
endocytosis and dynamin-:181/:1/3%A*+(0/%7(36%1/:0+0@1+N5%Cell 2009, 137(3), 433-444.
V*1+(/E,%VN%<NT%?@(36,%>N%KNT%Q-80/,%>N%uN%.I1E*2-3(0/%0A%@I!<%-99*@*2-3(0/%;4%-%K*@-/%
Immunodeficiency Virus trans-93('-30)%8)031(/N5%Cell 1987, 48, 691-701.
Pitt, S. W.; Majumdar, A.; Serganov, A.; Patel, D. J.; Al-Hashimi, H. M. "Argininamide
binding arrests global motions in HIV-1 TAR RNA: comparison with Mg2+-induced
conformational stabilization." J. Mol. Biol. 2004, 338(1), 7-16.
d)(91,%HN%gNT%a)9-2,%!NT%!-p-0p1,%INT%$-/p+,%RN%<N%.K=L-1 viral proteins gp120 and Tat
induce oxidative +3)1++%(/%;)-(/%1/:03612(-2%9122+N5%Brain Res. 2005, 104, 557-63.
d*AA1),%aN%$N,%d0/3)1220,%uN%DNT%%K0221/;19p,%uN%uNT%D(/p,%uN%<NT%D(1++2(/E,%GN%GN%.<93('-3(/E%$%
Q122%?(E/-2(/E%7(36%>1A(/1:%V*23('-21/3%G(E-/:+N5%ACS Chem. Biol. 2007, 2(4) 252-
262.

%..#
#
R-/Y-/%>-+,%?NT%u-@112,%?N%.$(020E4%0A%361%K=L%!1A%8)031(/5%Indian. J. Med. Res. 2005, 121,
315-332.
Rusnati, M.; Urbinati, C.; Caputo, A.; Possati, L.; Lortat-Jacob, H.; Giacca, M.; Ribatti, D.;
d)1+3-,%VN%.d1/30+-/%d024+*2A-31%-+%-/%=/6(;(3(0)%0A%am3)-cellular HIV-&%H-3N5%J. of Biol. Chem. 2001, 276(25), 22420-22425.
?12;4,%VN%uNT%$-(/,%aN%?NT%G*9(7,%dNT%y%d131)2(/,%$N%VN%.?3)*93*)1,%+1i*1/91%-/:%80+(3(0/%0A%
the stem-loop in TAR determine transcriptional elongation by Tat through the HIV-1
long termin-2%)181-3N5%Genes Dev. 1989, 3, 547-558.
Singh, I. N.; Goody, R. J.; Dean, C.; Ahmad, N. M.; Lutz, S. E.; Knapp, P. E.; Nath,
<NT%K-*+1),%DN%qN%.<808303(9%:1-36%0A%+3)(-3-2%/1*)0/+%(/:*91:%;4%6*@-/%%%
immunodeficiency virus-1 Tat and gp120: Differential involvement of caspase-3
-/:%1/:0/*921-+1N5%%G . J. Neurovirol. 2004, 10, 141-151.
Sodroski, J.; Patarca, R.; Rosen, C.; Wong-?3--2,%qNT%K-+123(/1,%RN%<N%.G09-3(0/%0A%361%3)-/+-
acting region on the genome of human T-9122%24@8603)08(9%'()*+%3481%===N5%Science
1995a, 229, 74-77.
Sodroski, J. G.; Rosen, C. A.; Wong-Staal, F.; Salahuddin, S. Z.; Popovic, M.; Arya, S.;
M-220,%IN%QNT%K-+123(/1,%RN%<N%.H)-/+-acting transcriptional regulation of human T-
cell leukemia virus 3481%===%20/E%31)@(/-2%)181-3N5%Science 1995b, 227, 171-173.
?3)0/E,%GN%aNT%-/:%D(1++2(/E,%GNGN%.<%M1/1)-2%?4/3613(9%I0*31%30%>1A(/1:,%Biologically
<93('1%V*23('-21/3%<))-4+N5%J. Am. Chem. Soc. 1999, 121(26), 6193-6196.
Tamilarasu, N.; Huq, I.; Rana,%HNVN%.H-)E13(/E%I!<%7(36%d183(:0@(@13(9%g2(E0@1)+%(/%
K*@-/%Q122+N5%Bioorg. Med. Chem. Lett. 2001, 11, 505-507.
H4-E(,%VNT%I*+/-3(,%VNT%d)1+3-,%VNT%M(-99-,%VN%.=/31)/-2(\-3(0/%0A%K=L-1 Tat requires cell
+*)A-91%618-)-/%+*2A-31%8)0310E249-/+N5%J. Biol. Chem. 2001, 276, 3254-3261.
UNAIDS .M20;-2%I180)3^%UNAIDS )180)3%0/%361%E20;-2%-(:+%18(:1@(9%`U&`N5%WHO, 2012;
pp 1-110.
[);(/-3(,%QNT%Q6(0:122(,%dNT%I*+/-3(,%VN%.d024-/(0/(9%>)*E+%-/:%L()-2%g/90E1/1+(+^%-%!0'12%
Approach to Control Infection, Tumor-assoc(-31:%=/A2-@@-3(0/%-/:%</E(0E1/1+(+N5%
Molecules 2008, 13, 2758-2785.
Vendeville, A.; Rayne, F.; Bonhoure, A.; Bettache, N.; Montcourrier, P.; Beaumelle, B.
.K=L-1 Tat enters T cells using coated pits before translocating from acidified
endosomes and elic(3(/E%;(020E(9-2%)1+80/+1+N5%Mol. Biol. Cell. 2004, 15, 2347-2360.
Wang, D.; Iera, J.; Baker, H.; Hogan, P.; Ptak, R.; Yang, L.; Hartman, T.; Buckheit, Jr., R.
W.; Desjardins, A.; Yang, A.; Legault, P.; Yedavalli, V.; Jeang, K. T.; Apella, D. H.
.V*23('-21/3%$(/:(/E%g2(E0@1)+%=/6(;(3%K=L%H-3-TAR Interaction Critical for Viral
I182(9-3(0/N5 Bioorg. Med. Chem. Lett. 2009, 19(24), 6893]6897.
doi: 10.1016/j.bmcl.2009.10.078

%.%#
#
R-/E,%vNT%R-/E,%lNT%I-/-,%HN%VNT%.d)031(/%0)(1/3-3(0/%(/%361%H-3-TAR complex determined
;4%8+0)-21/%860309)0++2(/p(/EN5J. Biol. Chem. 1996, 271, 16995-
Weeks, K. M.; Ampe, C.; Scultz, S.QNT%?31)3\,%HN%<NT%Q)0361+,%>N%VN%.q)-E@1/3+%0A%361%K=L-1
3-3%8)031(/%+819(A(9-224%;(/:%H<I%I!<N5%Science 1990, 249, 1281-1285.
Weissman, J. D.; Brown, J. A.; Howcroft, K. T.; Hwang, J.; Chawla, A.; Roche, P. A.;
Schiltz, L.; Nakatani, Y.; Singer, D. S. .K=L-1 Tat binds TAFII250 and represses
TAFII250-:181/:1/3%3)-/+9)(83(/%0A%@-Y0)%6(+3090@8-3(;(2(34%92-++%=%E1/1+N5%Pro. Natl. Acad. Sci. 1998, 95, 11601-11606.
R1(\@-/,%KNT%H0),%nN%.H-)E13(/E%I!<%7(36%?@-22%V0219*21+N5%ChemBiochem 2003, 4, 998-
1007.
Ya/E,%VN%.>(+90'1)(1+%0A%H-3-TAR Interaction Inhibitors for HIV-&N5%Curr. Drug Targets Infect Disord. 2005, 5(4), 433-444.
v6-0,%KNT%G(,%uNT%u(-/E,%GN%.=/6(;(3(0/%0A%K=L-1 TAR RNA ] Tat peptide complexation using
8024C-9)42(9%-9(:BN5%Biochem. Biophys. Res. Commun. 2004, 320 (1), 95-99.

%.&#
#
Appendix: 1H N M R Spectra:
%.'#
#
!
#
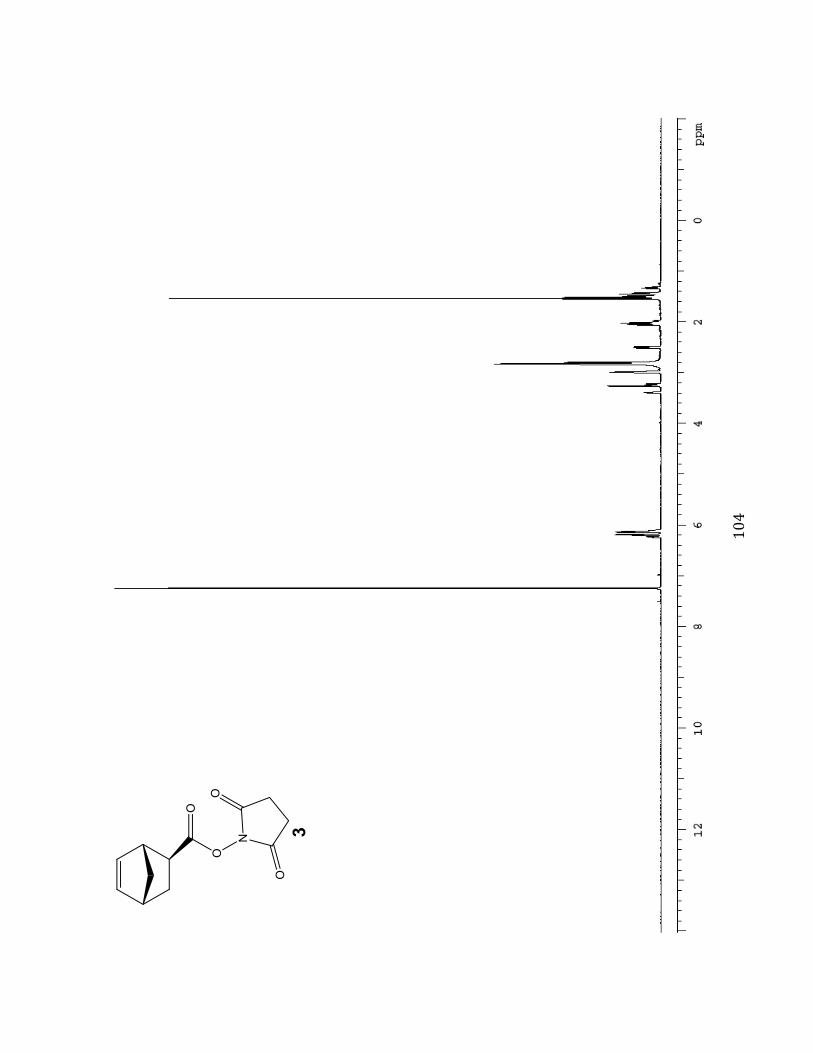
%.(#
#
!
#
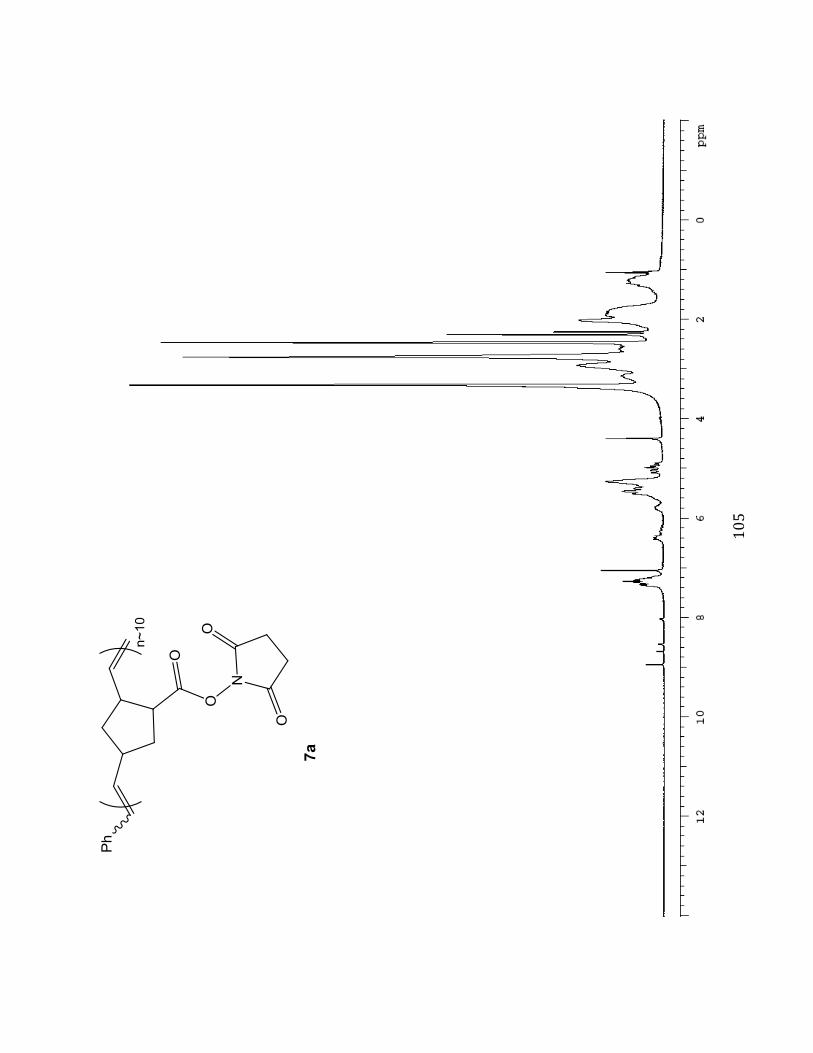
%.)#
#
!
#
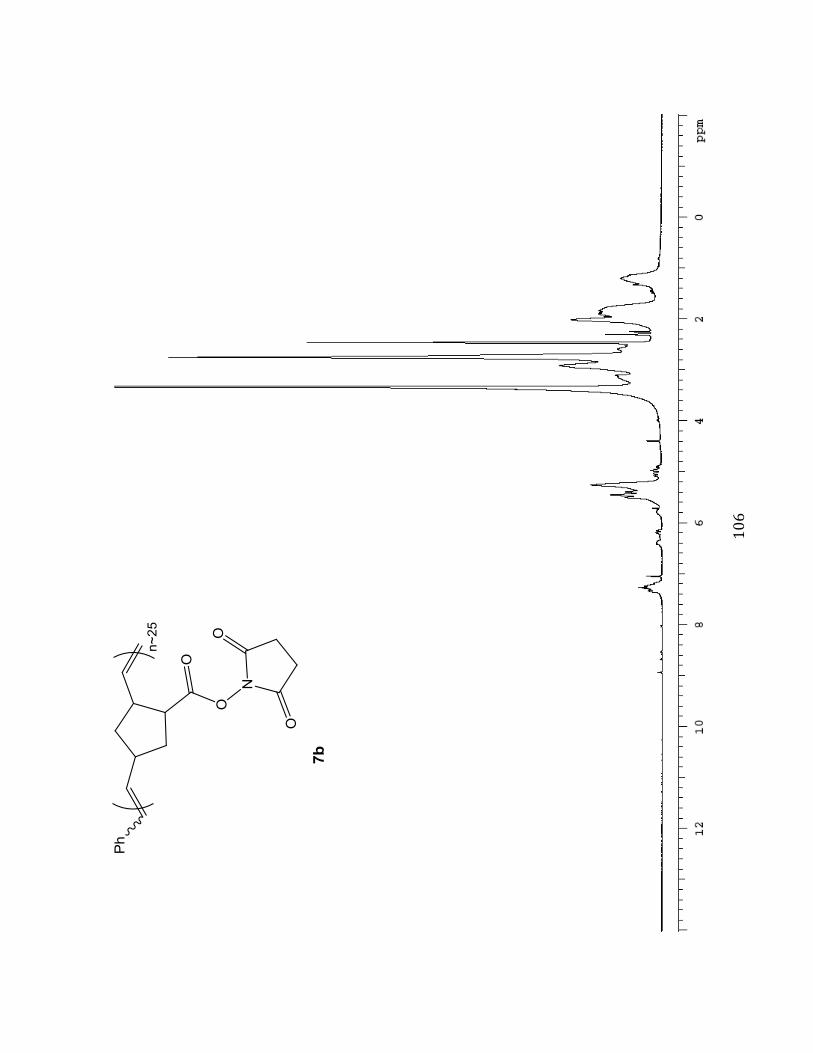
%.*#
#
!!
#
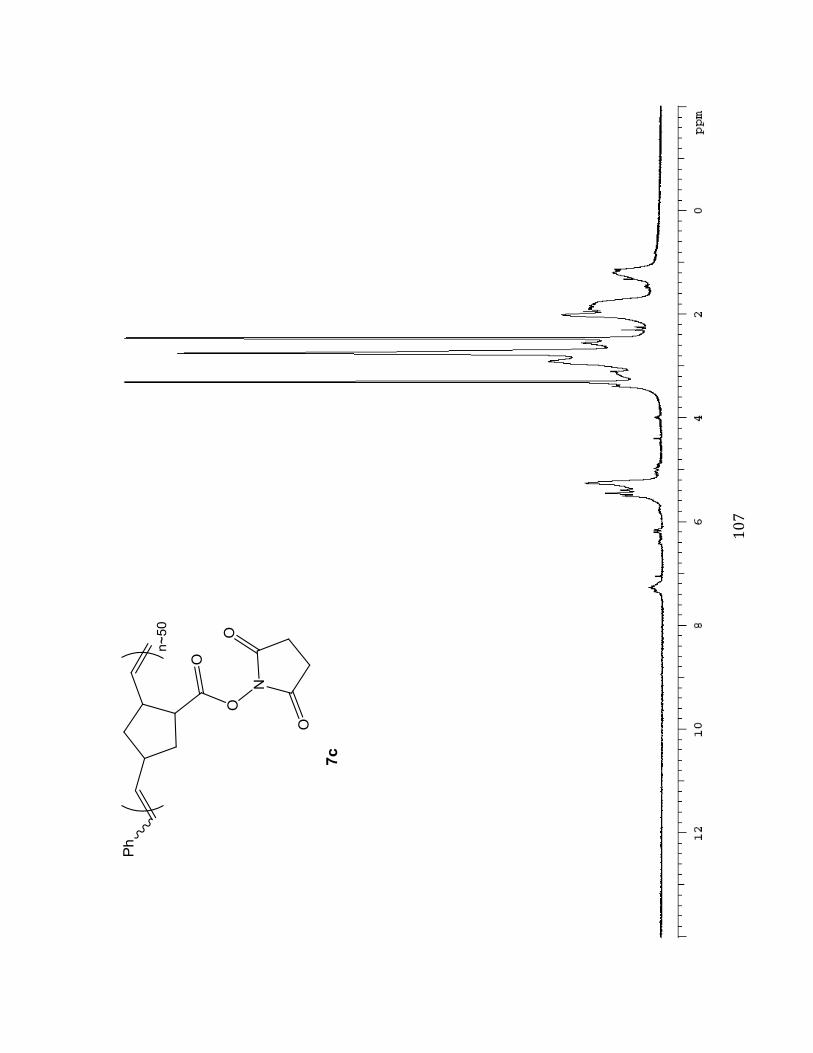
%.+#
#
!
#
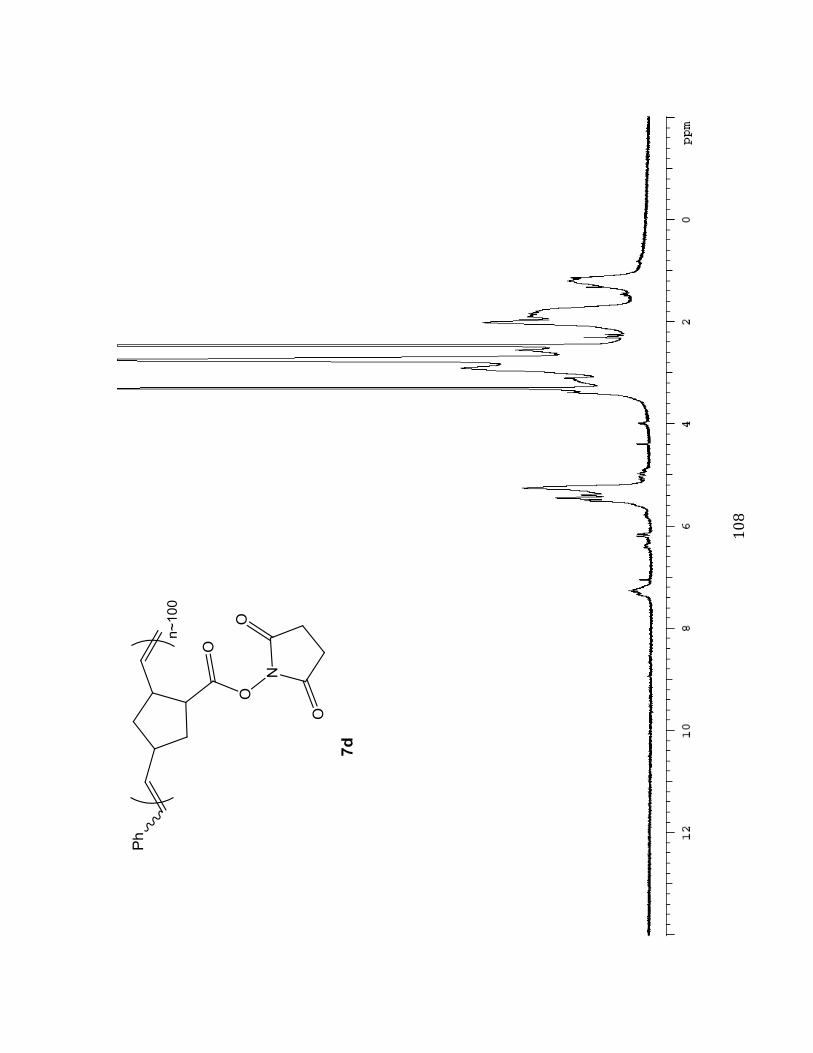
%.,#
#
!
#
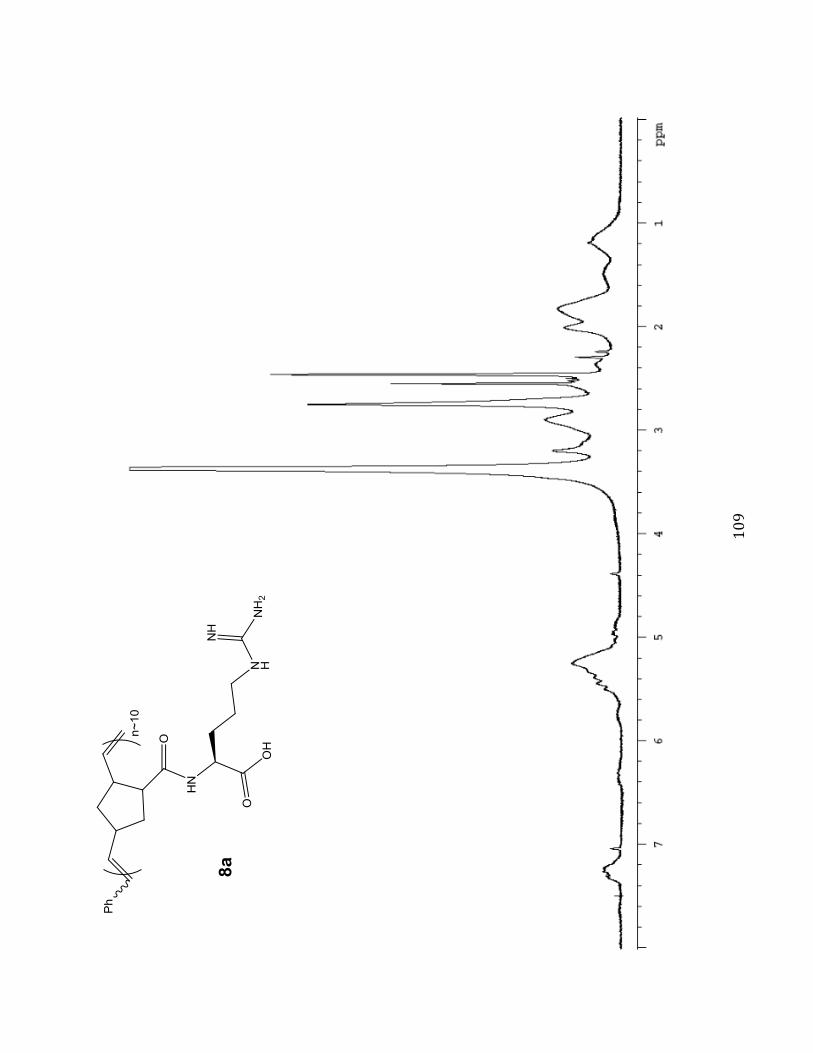
%.-#
#
!
#
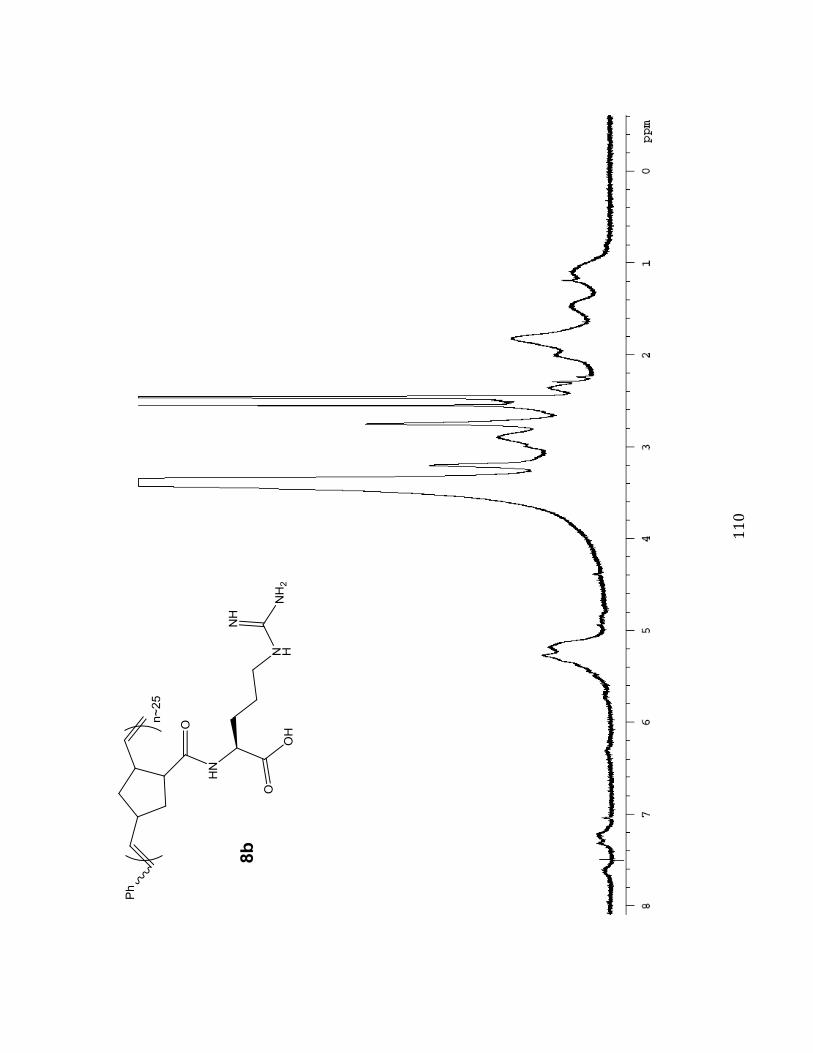
%%.#
#
!
!
#
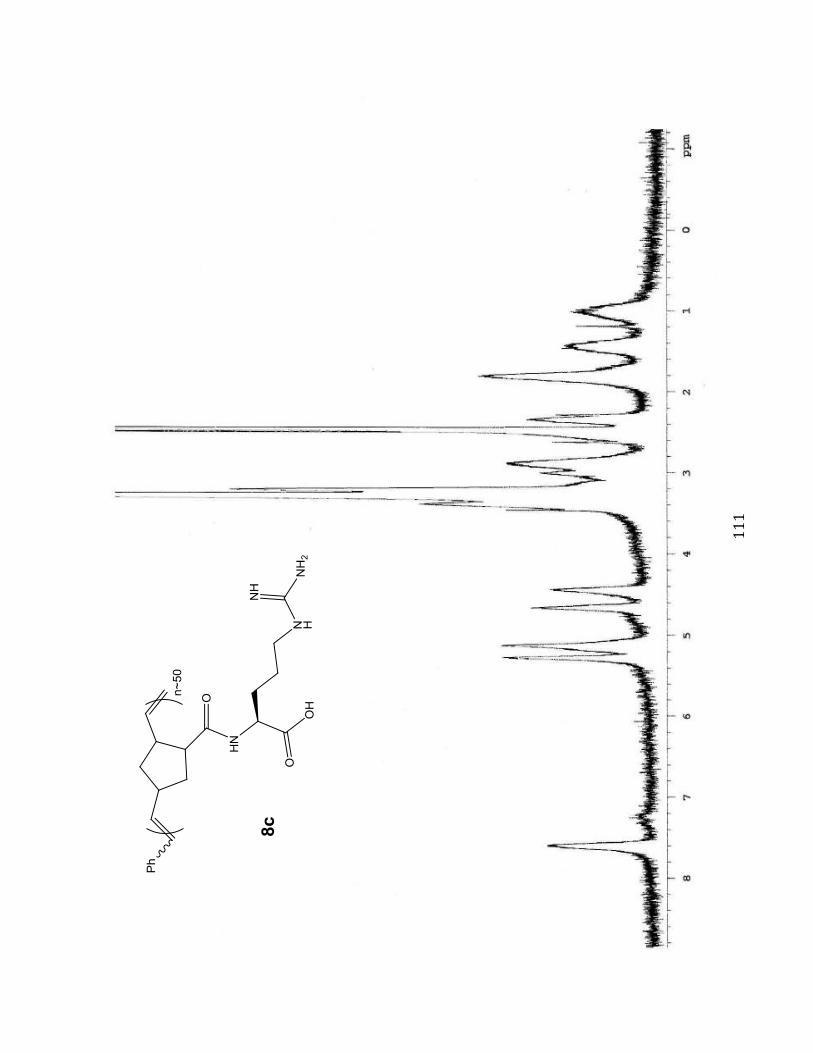
%%%#
#
!
#

%%&#
#!!
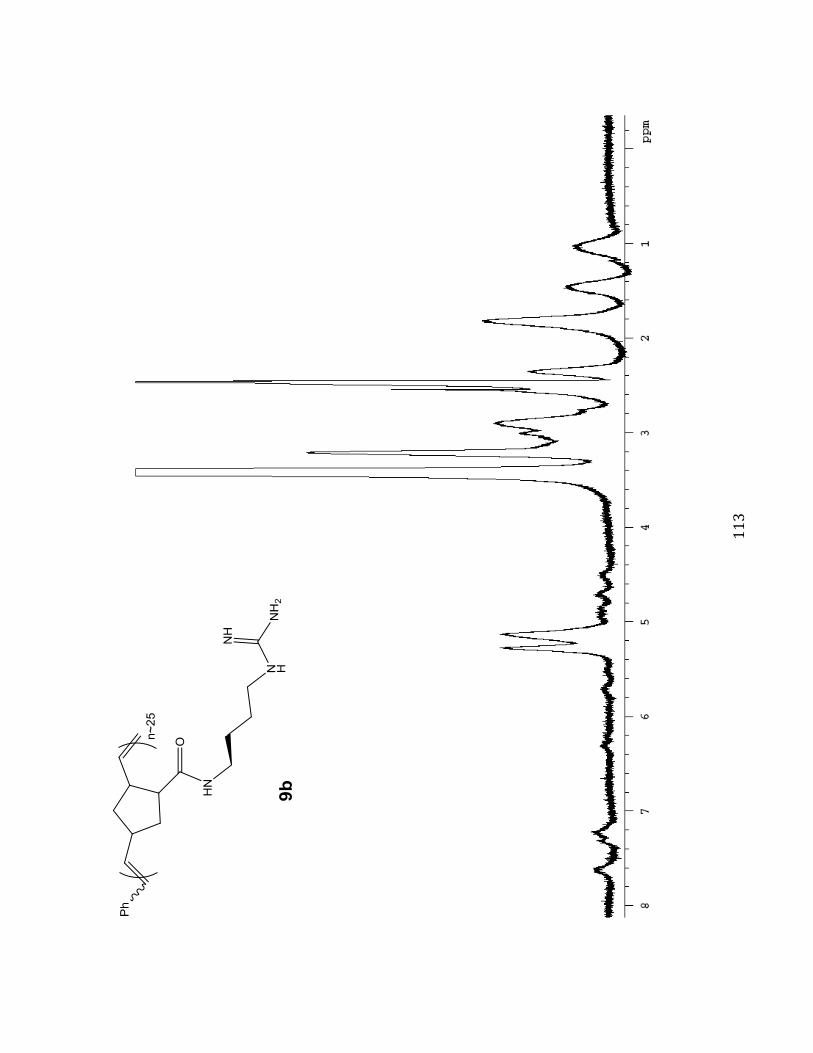
%%'#
#
!
!
#
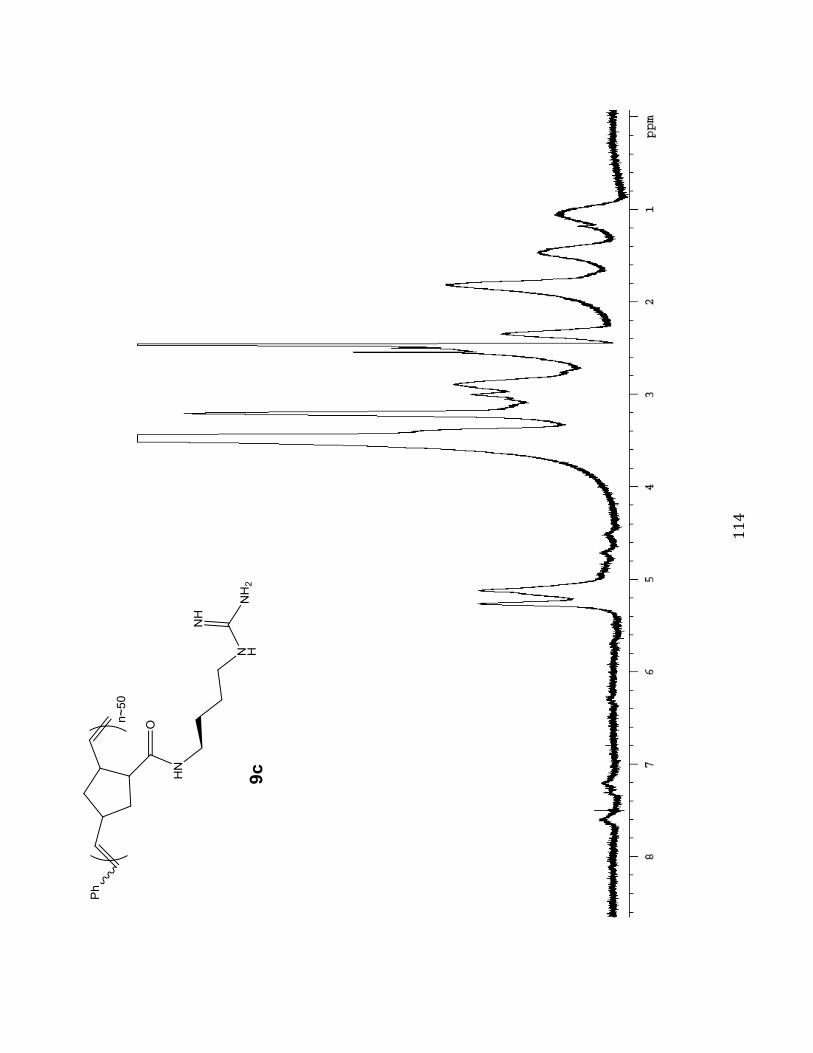
%%(#
#
!
#
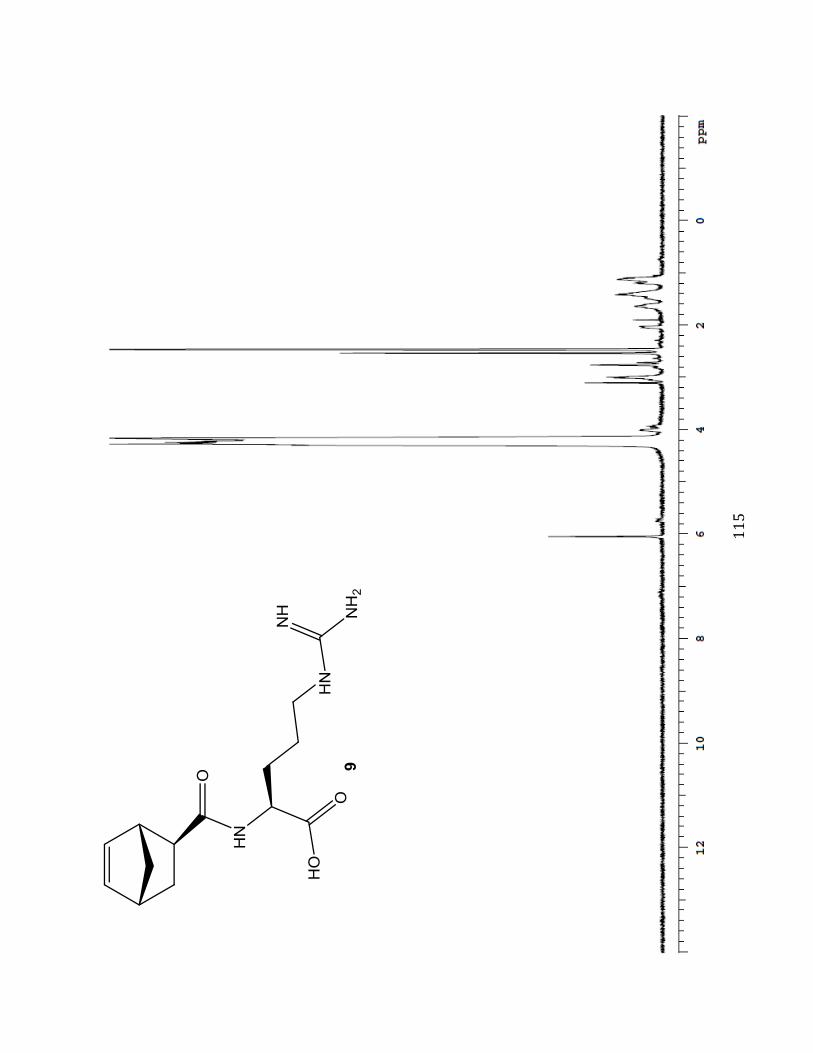
%%)#
#
#