a manuscript submitted in partial fulfillment of
TRANSCRIPT

The Evaluation of Polycythemia and the Diagnosis of Polycythemia Vera
By
Kerri Lantz Petersen
A manuscript submitted in partial fulfillment of the requirements for the degree of
MASTER OF NURSING
WASIDNGTON STATE UNIVERSITY College of Nursing Education
May 2001
..

To the faculty of Washington State University:
The members of the committee appointed to examine the College ofNursing
Education Research requirements and manuscript of Kerri Lantz Petersen
find it satisfactory and recommend that it be accepted.
~lrq~ C air: Dr. Lorna Schumann
Dr. Billie Severtsen
~/V~~t:=-. Naomi Lungst1'OlIlRN, MN
ii

TABLE OF CONTENTS
page
ABSTRACT vi
LIST OFTABLES iv
LIST OFFIGURES v
rnTRODUCTION -1
EPIDEMIOLOGY 1
ETIOLOGy 2
PATHOPHYSIOLOGY 2
CLOOCAL COURSE 3
CLrnICAL PRESENTATION 4
LABORATORY FEATURES 6
DIFFERENTIAL DIAGNOSIS 7
CLrnICAL DECISION MAKING 10
TREATMENT 14
COMPLICATIONS 17
CONCLUSION 18
REFERENCES 19
iii

LIST OF TABLES
1. The Occurrence of Signs and Symptoms in Patients 30
2. Differential Diagnosis of Polycythemia Vera 31
3. Laboratory Differentiation of Polycythemia 32
4. PVSG Criteria for the Diagnosis of Polycythemia Vera 33
5. The Rotterdam Criteria of Polycythemia Vera 34
6. Proposed Modified Criteria for the Diagnosis of Polycythemia Vera 35
7. Laboratory and Diagnostic Tests for Polycythemia Vera 36
8. Patient Education Resources 42
iv

LIST OF FIGURES
1. Diagnostic Approach to "Suspected Polycythemia Vera" 43
2. Diagnostic Approach to an Elevated Hematocrit 44
v

The Evaluation of Polycythemia and the Diagnosis of Polycythemia Vera
Abstract
By Kerri Lantz Petersen Washington State University
May,2001
Chair: Dr. Lorna Schumann
Polycythemia vera (PV) is a rare clonal blood disorder marked by an elevated hematocrit,
hemoglobin, and red cell mass. Although the etiology is unknown, PV is characterized by the
proliferation of hematopoietic progenitor cells which creates an increased red cell mass
accompanied by an expanded blood volume. Red blood cell production continues independent of
increased blood viscosity. Onset is insidious and the patient may present without symptoms or
complain of headache, dizziness, or pruritus. Physical examination typically reveals
splenomegaly, plethora, and retinal vein engorgement. Laboratory features include leukocytosis,
thrombocytosis, and erythrocytosis. Serum erythropoietin levels are low or absent. PV must be
distinguished from relative and secondary polycythemia. Relative polycythemia causes a
moderate increase in hematocrit frequently secondary to fluid contraction. The red cell mass,
although not truly increased, reflects hemoconcentration. Secondary polycythemia is suspected,
if an elevated hematocrit occurs without leukocytosis, thrombocytosis, and splenomegaly. The
red cell mass measurement and serum erythropoietin level will be increased. Tests used in the
differential diagnosis of PV include red cell mass determination, erythropoietin assays, arterial
oxygen saturation, and renal ultrasounds. Regular phlebotomy is the safest treatment for PV.
vi

Myelosuppressive agents may be given in addition to phlebotomy therapy. The median survival
time for patients with PV is 11 to 15 years with appropriate medical treatment.
vii

The Evaluation of Polycythemia and the Diagnosis of Polycythemia Vera
Polycythemia vera (PV) is a relatively rare clonal blood disorder often discovered
incidentally through routine blood testing (Wasserman, 1954; Adamson, Fialkow, Murphy,
Prchal, & Steinmann, 1976; Talarico, 1998). The complete blood count characteristically shows
an elevated hematocrit and hemoglobin with an increased red cell count (Peterson & Wasserman,
1995; Ferri, 2001). PV must be distinguished from other causes of polycythemia, including
relative and secondary polycythemia (Spivak, 2001). How does the primary care provider
accurately evaluate an elevated hematocrit and hemoglobin and the polycythemias? It has been
reported that 50% of persons with untreated PV die within the first 18 months after the first signs
or symptoms appear (Chievitz & Thiede, 1962). Diagnosis, careful monitoring, and appropriate
treatment can reduce the risk of complications (Spivak, 2001).
Epidemiology
The reported incidence of PV has varied greatly due to a number of factors including
quality of medical care, diligence of record keeping, and the age, ethnic, and racial distributions
of study populations (Modan, 1995; Michiels et aI., 2000; Chaiter, Brenner, Aghai, & Tatarsky,
1992; Berglund & Zettervall, 1992). Prochazka and Markowe (1986), in their large study of
incidence rates from New Wales and England, found a rate of 5 to 10 cases per million per year
with a male to female ratio of 1:3. Chaiter et aI., (1992) found a slight increase in males as
opposed to females. Increased incidences have occurred among those of Jewish origin with 11.4
cases per one million residents in northern Israel with myeloproliferative disorders in general
occurring with a higher incidence among Ashkenazi Jews specifically (Chaiter et aI., 1992). The
median age at onset is 56.9 years (Modan & Lilienfeld, 1965). Rare cases have occurred during

2
childhood (Marlowe & Fairbanks, 1960; Aggeler, Pollycove, Hoag, Donald, & Lawrence, 1961).
Exposure to radiation has been associated with an increased incidence of PV (Caldwell, Kelley,
Heath, & Zack, 1984).
Etiology
Although the etiology is unknown, this disorder is marked by an autonomous clonal
proliferation of bone marrow progenitors, such as erythroid, granulocytic, and megakaryocytic
cells representing a neoplastic process (Adamson et aI., 1976). Stem cell cloning and
proliferation of red blood cells (RBCs) in patients with PV occurs independent of erythropoietin
production as demonstrated by bone marrow and peripheral blood in vitro culture studies
(Zanjani & Lutton, 1977; Prchal, Adamson, Murphy, Steimann, & Fialkow, 1978). Recent
studies to explain the erythropoietin- independent proliferation of red blood cells have not shown
a functional or structural abnormality in the erythropoietin receptor (Hess et aI., 1994;
Asimakopoulos et aI., 1997). Recently, Silva and colleagues (1998) examined the role of a Bcl-2
gene, an apoptosis-inhibiting oncoprotein, and its possible relationship to the erythropoietin
independent proliferation of red blood cells (RBCs). Molitemo, Hankins, and Spivak (1998)
found that the thrombopoietin receptor, Mpl, on platelets and megakaryocytes was absent or
greatly reduced in their patients with PV. Studies suggest a defect in programmed cell death and
proliferation independent from growth factors (Silva et aI., 1998; Molitemo et aI., 1998).
Pathophysiology
Polycythemia is defined as an increase in packed cell volume (hematocrit). The increased
hematocrit may occur with an increased red cell mass (secondary polycythemia and PV) or a
normal red cell mass (RCM) (Pearson & Messinezy, 1996). The increased RCM can be due to a
physiologic appropriate or inappropriate erythropoietin response. Erythropoietin, produced

3
mainly by the kidneys in adults, normally regulates erythropoiesis (proliferation and maturation
of erythryoid progenitor cells). Erythropoietin is a potent glycoprotein carefully controlled by the
body to maintain stability. Hypoxia can stimulate the production of erythropoietin, producing red
blood cells to maintain tissue oxygenation by increasing the number of oxygen carrying cells in
the blood. The plasma erythropoietin level (EPa) reflects tissue oxygenation. Tumors capable
of causing inappropriate erythropoietin production can elevate EPa levels in spite of normal
tissue oxygenation (Waldmann, Rosse, & Swarm, 1968; Balcerak & Bromberg, 1975). PV is
characterized by neoplastic progenitor growth and erythroid colony forming units (CFU-E),
although EPa production and concentration are decreased and arterial oxygen saturation is
normal. The proliferation of RBCs continues independently of exogenous erythropoietin
stimulation. CFU-E have been shown to form in marrow cell culture without the addition of
exogenous erythropoietin in patients with PV (Weinberg et aI., 1989; Prchal & Axelrad, 1974;
Zanjani, Lutton, Hoffman, & Wasserman, 1977; Eaves & Eaves, 1978). London, Shemin, West,
and Rittenberg (1949) demonstrated a two and one-half fold increase in red cell production by
the hyperplastic marrow of PV patients. The elevated RCM (equal to or greater than 36 mUkg
for males, equal to or greater than 32 mUkg for females) that results from this proliferation
causes increased whole-blood viscosity (Wasserman, 1954; Shien & Gallik, 1995).
Clinical Course
PV is characterized by: the preerythrocytic phase; the erythrocytic phase; the proliferative
phase, and the "spent" or postpolycythemic phase. During the preerythrocytic phase, which can
last many years, the patient frequently remains asymptomatic. The RBC mass slightly increases
and the patient may complain of pruritus after bathing. The only sign noted during the physical
exam may be splenomegaly. The erythrocytic phase presents with ischemic events due to

4
increased blood viscosity and RCM. Bleeding episodes, cyanosis, plethora, and an elevated
platelet count can occur. Treatment may prolong the erythrocytic phase for 5 to 20 years. The
proliferative phase is characterized by bruising, hemorrhage, leukocytosis, and progressive
splenomegaly. Ten years may pass between phases 3 and 4. Approximately 20% of patients
reach the "spent" phase and may experience severe anemia with progression to leukemia or bone
marrow fibrosis (David, 1995). Transfusions may be required during this phase (Beutler, 2001).
Mortality is high once the patient reaches the fourth phase (David, 1995).
Wasserman and Peterson (1995) describe the asymptomatic developmental phase of PV
as insidious and possibly accompanied by hepatosplenomegaly, thrombocytosis, or an elevated
hemoglobin without an increase in the red cell mass. The next phase, the erythrocytic phase,
may last from 5 to 20 years. Laboratory results show an increased red cell mass, with an
expanded blood volume and the proliferation of all the elements of the bone marrow,
panmyelosis. The following phase, known as the "spent" or "stable" phase, is characterized by
postpolycythemic myeloid metaplasia. This phase of thrombocytopenia, leukopenia, and anemia
is accompanied by a stabilization of the red cell mass and blood volume. The bone marrow may
become fibrotic and patients may succumb or progress to acute leukemia (Wasserman, 1954;
Peterson & Wasserman, 1995; Silverstein, Brown, & Linman, 1973; Silverstein, 1974).
Clinical Presentation
The onset of PV is insidious and its course progressive. The patient may be
asymptomatic or experience signs and symptoms related to increased blood viscosity and
expanded blood volume (Peterson & Wasserman, 1995; Berk, 1975). Initial presentation varies
from patient to patient. Family members may have expressed concerned about the patients ruddy
comple.xion and hypertension may have been found during the physical examination (Berlin,

5
1975, 1995). Initial signs and symptoms may include headache, peptic ulcer, tinnitus,
thrombosis, dizziness, paresthesias, gastrointestinal bleeding, intermittent claudication, gout, or
pruritus (Berlin, 1975, 1995; Peterson & Wasserman, 1995; Beutler, 2001). Generalized pruritus
is intense and exacerbated by warm baths and showers. The cellular release of histamine from
increased basophils may be responsible for the intense itching although antihistamines are
usually ineffective (Krajink & Zylicz, 2001). Other symptoms include visual disturbances and
abdominal discomfort (Berlin, 1975; Hocking, 1995). Weight loss may be apparent (Wasserman,
1954; Berlin, 1975; Talarico, 1998).
Physical examination often reveals cyanosis, plethora, splenomegaly, congestion of oral
mucosa, and retinal vein engorgement (Berlin, 1975; Talarico, 1998; Ferri, 2001). Episodes of
epistaxis, bruising, and gingival bleeding can occur. Small vessel insufficiency may result in
cyanosis and even frank gangrene of the toes and fingers (Talarico, 1998). Erythromelalgia,
redness and burning pain in the extremities, may be a presenting symptom of PV (Michiels et aI.,
1985). Transient ischemic attacks, cerebrovascular accidents, pulmonary emboli, arterial
thrombosis, or deep vein thrombosis can also be present initially or during the course of the
disease (Peterson & Wasserman, 1995). The Gruppo Italiano Studio Policitemia (1995)
conducted a retrospective study that revealed 140/0 of PV patients had a thrombotic event prior to
diagnosis. Hyperviscosity of the blood with an elevated hematocrit is responsible for many of
the features of PV especially those related to the brain, lungs, and retina (Peterson & Wasserman,
1995; Chien & Gallick, 1995). Table 1 lists signs and symptoms characteristic of PV.

6
Laboratory Features
The hallmarks of PV include erythrocytosis, thrombocytosis, and leukocytosis. The
platelet count is often above 400 x 109/L and the white cell count can exceed 12 x 109/L
(Wasserman, 1954; Berlin, 1966, 1975, 1995). Mild basophilia may be present (Berlin, 1966,
1995). The hematocrit will be elevated, and may be above 60%, although microcytosis with iron
deficiency can result in a moderately increased hematocrit (Fairbanks & Tefferi, 2000;
Silverstein & Tefferi, 2000). Erythrocytosis occurs in other forms of polycythemia. The RCM,
in PV patients will be more than 25% above what is predicted for the patient's height and weight
(Messinezy & Pearson, 1999). The RCM increase contributes to an increase in blood viscosity.
The erythrocytosis and increased RCM occurs without a normal erythropoietin response. The
serum EPO level is decreased or undetectable (Napier & Wieczorek, 1981; Erslev, Caro, Kansu,
Miller, & Cobbs, 1979; de Klerk et aI., 1981). Occasionally the EPO level will remain normal
(Cotes et aI., 1986). Elevated EPO levels raise the suspicion of secondary polycythemia due to
hypoxemia (Tefferi, 1999; Messinezy & Pearson, 1999).
Arterial oxygen saturation is usually above 92% unless the patient has superimposed lung
disease such as chronic bronchitis or emphysema. The Polycythemia Vera Study Group (PVSG)
found that 10% of their patients demonstrated blood saturation levels between 88 and 92% and a
smaller number had unexplained levels below 880/0 (Berlin, 1966, 1975, 1995).
Nonspecific laboratory results include an elevated leukocyte alkaline phosphorus (LAP)
above 100 units, an elevated reticulocyte count above 1.5%, hyperuricemia, and an increased
serum vitamin B12 or B12-binding capacity (Berlin, 1966, 1975, 1995; Wasserman, 1954;
Tefferi, 1998). Coagulation tests are usually normal (Murphy, Davis, Walsh, and Gardner, 1978;
HoclGng, 1995).

7
Bone marrow biopsies from patients with PV have been studied extensively since 1967
by the Polycythemia Vera Study Group (PVSG). They found that the marrow of untreated PV
patients demonstrated panhyperplasia, with wide individual variation. Reticulin fibrosis has also
been found after treatment and during the spent phase of PV. The marrow cavity may become
"obliterated" by fibroblastic or osteoblastic proliferation and the extraosseous marrow production
of blood cells occurs (myeloid metaplasia) (Wasserman, 1954; Peterson & Ellis, 1995).
Differential Diagnosis
The cause of an elevated hematocrit can, in most cases, be determined by the patient's
history, a thorough family history, physical examination, complete blood count, and chest x-ray
(Landaw, 1996). Previous laboratory results should be made available for comparison to recent
laboratory findings. Polycythemia vera must be distinguished from secondary polycythemia, and
relative, or "spurious" polycythemia since all three cause an elevated hematocrit and hemoglobin
(Copelan & Balcerzak, 1995; Weinreb, 1995; Spivak, 2001). Table 2 lists the various causes of
polycythemia.
Types of Polycythemia
The actual incidence of relative polycythemia, also referred to as "spurious", "stress", or
Gaisbock's polycythemia, remains unknown (Weinreb, 1995). Patients with relative
polycythemia are generally younger than those found to have PV (Weinreb & Shih, 1975).
Relative polycythemia is not a true erythrocytosis. It is characterized by a moderate increase in
the hematocrit (Berlin, 1966). The hematocrit ranges consistently above 49% and the erythrocyte
count is elevated. The red cell mass remains normal, but may be in the upper normal range.
Plasma volume may be normal or reduced (Weinreb, 1995). The platelet, white blood cell, and
reticulocyte counts are usually normal (Berlin, 1966; Weinreb & Shih, 1975). Symptoms include

8
epigastric discomfort, lethargy, fatigue, sweating, palpitations, lightheadedness, headache,
dyspnea, and dizziness. Pruritus and weight loss are uncommon. Physical examination may
reveal plethora and conjunctival suffusion without palpable splenomegaly. Hypertension and
obesity are common findings (Burge, Johnson, & Prankerd, 1975; Weinreb & Shih, 1975). The
patient's history may reveal common causes of dehydration such as febrile illness, vomiting, or
diarrhea (Lopez, 1996). The medication history may include diuretics, excessive caffeine intake,
and cardiac medications such as calcium channel blockers and beta-blockers (Burge, Johnson, &
Prankerd, 1975; Landaw, 1996).
Weinreb (1995) classifies relative polycythemia as acute or transient and chronic or
sustained polycythemia. Transient polycythemia most commoflly occurs with a contracted fluid
volume caused by dehydration. The dehydration may be caused by vomiting, diarrhea, or
insensible fluid losses, but is usually corrected with medical treatment. Chronic relative
polycythemia can occur in the context of a normal or low-normal plasma volume with a high
normal RCM (normovolemic relative polycythemia) or a normal RCM with hemoconcentration
secondary to a reduced plasma volume (hypovolemic relative polycythemia). A normal or low
nonnal plasma volume with a high-normal RCM may be idiopathic or due to intermittent
hypoxemia. Smoking, sleep apnea, and postural hypoxemia have been linked to elevated
hematocrits (Smith & Landaw, 1978; Watts & Lewis, 1983; Weinreb, 1995). Idiopathic cases of
nonnovolemic polycythemia can be viewed as normal variants (Brown, Gilbert, & Krauss, and
Wasserman, 1971). Both hypovolemic and normovolemic polycythemias may be associated with
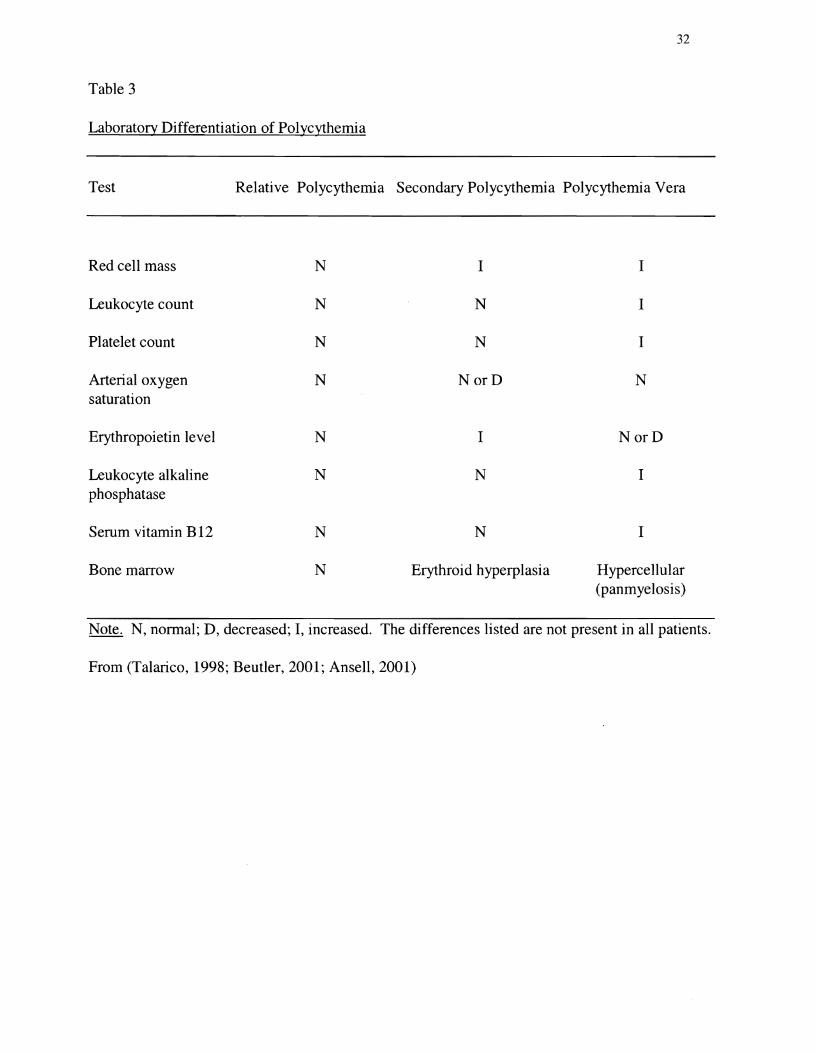
obesity, smoking, hypertension, and/or sleep apnea (Weinreb, 1995). Table 3 lists typical
laboratory findings in the differential diagnosis of polycythemia.

9
Secondary polycythemia (a nonclonal disorder) should be suspected when the hematocrit,
hemoglobin, and RBC count are elevated. Thrombocytosis, leukocytosis, and panhyperplasia of
the bone marrow are found less often as opposed to PV (Balcerak & Bromberg, 1975). The
ReM will be increased (Balcerak & Bromberg, 1975; Copelan & Balcerak, 1995). Patients
characteristically present without splenomegaly unless metastatic neoplasms occur
simultaneously (Ferri, 2001). Arterial oxygen saturation is reduced if a chronic hypoxic
condition is present (Balcerak & Bromberg, 1975). The EPO concentration will be increased
representing a physiologically appropriate or inappropriate response (Copelan & Balcerak, 1995).
Secondary polycythemia caused by tissue hypoxia, such as chronic pulmonary disease,
cardiovascular shunts, and high altitudes, can be noted from the history, physical examination,
and laboratory tests (Berlin, 1966; Balcerak & Bromberg, 1975; Copelan & Balcerak, 1995;
Lopez, 1996). Rare hemoglobinopathies associated with high oxygen affinity occasionally cause
secondary polycythemia reflected by abnormal oxyhemoglobin dissociation curves. Ectopic
production of erythropoietin can occur with renal carcinoma, uterine tumors, or hepatoma, and
the erythropoietin level will be elevated with a normal arterial oxygen saturation. Renal lesions
can also cause secondary polycythemia with increased erythropoietin production (Berlin, 1975,
1995; Copelan & Balcerak, 1995; Waldmann et aI., 1968).
Secondary polycythemia due to carbon monoxide also results in an elevated hematocrit
from an increased circulating carboxyhemoglobin level. It is important to question the patient
about possible chronic exposure to carbon monoxide from faulty heating systems or automobile
exhaust. Patients should be questioned about cigar, cigarette, and marijuana use (Landaw, 1998).
Inhaled carbon monoxide affects the release of oxygen from hemoglobin, leads to the formation
of carboxyhemoglobin, and stimulates erythropoietin production (Copelan & Balcerak, 1995).

10
The serum EPO level rises while plasma volume decreases. Carboxyhemoglobin measurements
should be timed to coincide with peak levels. For example, blood should be drawn in the early
morning, if a faulty home heating system is suspected (Landaw, 1996).
Clinical Decision Making
No single diagnostic marker exists to diagnose PV. Historically the PVSG diagnostic
criteria have been used to diagnose PV (Berk, 1997). Major and minor criteria are presented in
Table 4. RCM measurement with chromium-51 tagged RBCs has been recommended for the
initial evaluation of polycythemia to distinguish relative polycythemia from secondary
polycythemia and PV (Najean, Dresch, Rain, & Chomienne, 1995; Peereboom, 1996). RCM
determination helps confirm the diagnosis of PV, but does not distinguish between secondary
polycythemia and PV (Beutler, 2001; Lamy et aI., 1997). Differentiating secondary polycythemia
from PV has, until recently, relied on demonstrating pancytopenia in the peripheral blood,
increased RCM, palpable splenomegaly, arterial oxygen saturation, serum B12 binding capacity,
hemoglobin electrophoresis, and other tests to rule out causes of inappropriate erythropoietin
production (Modan & Lilienfeld, 1965; Berlin, 1975). The expense and inaccurate
interpretations of RCM determination coupled with the availability of more accurate tests has led
to various proposals to modify the original criteria designed by the PVSG (Michiels et aI., 1999;
Michiels & Juvonen, 1997; Tefferi, Solberg, & Silverstein, 2000; Messinezy & Pearson, 1999).
The Thrombocythemia Vera Study Group (TVSG) from The Netherlands has proposed a
revision of the PVSG criteria (See Table 5). The TVSG recommends replacing palpable
splenomegaly with bone marrow biopsy histopathology to improve the present PVSG criteria.
Michiels and Juvonen (1997) cite Kumick and colleagues' (1972) study of 90 bone marrow
specimens. Kumick et aI., found that 900/0 of PV patients had increased megakaryocytes and/or

11
eosinophils compared with 25% from the secondary polycythemia group. They concluded that
bone marrow sections should be included as a major criterion for PV diagnosis. Those patients
with anemia and hemorrhagic PV and those with splenomegaly secondary to an erythropoietin
producing tumor would be more accurately diagnosed, if bone marrow histopathology were
included in the criteria. The proposed criteria would aid the diagnosis of patients with secondary
polycythemia or early stage PV. Michiels and Juvonen (1997) propose using ultrasound or
computer tomography to assess more accurately spleen size, as opposed to only palpation of the
spleen. The Rotterdam criteria include erythroid colony formation in the absence of
erythropoietin. This occurs almost exclusively in PV (Michiels & Juvonen, 1997).
Pearson and Messinezy (1999) reevaluated the PVSG criteria with the newer diagnostic
tests available. See table 6 for Pearson and Messinezy's proposed diagnostic criteria. They
recommend eliminating the RCM measurement expressed as mL per kilogram, replacing it with
the more accurate recommendations from the International Council for Standardization
Hematology based on height and weight (Pearson, 2001; Pearson, 1995; Pearson & Messinezy,
1999). Pearson & Messinezy (1999) do not include bone marrow histopathology among their
proposed criteria due to the lack of "objective, quantitative methods of describing appearances"
to reduce sampling error histology. Clonality marker and palpable splenomegaly remain major
criterion (Pearson, 2001). Pearson (2001) recommends erythropoietin level and endogenous
erythroid colonies (EEC) remain minor criterion. He cites the expense of demonstrating
endogenous erythroid colony growth (EEC) and its low specificity for PV to justify this
recommendation.
Tefferi (1999) has suggested the use of serum EPO level and EEC assay as an alternative
approach to the diagnosis of PV (Figure 1). The presence or absence of PV-related features with

12
a normal serum EPO helps determine the diagnostic pathway. PV-related features are persistent
leukocytosis, persistent thrombocytosis, microcytosis, splenomegaly, generalized pruritus after
bathing, unusual thrombosis, and erythromelalgia (acral dysesthesia and erythema). Tefferi
claims that RCM measurement is expensive and may not identify cases of early PV or PV with
iron deficiency. Tefferi suggests the "narrow" reference range for RCM may result in a segment
of the normal population mistakenly diagnosed with PV. Cameskog et aI., (1999) have also
demonstrated the value of plasma EPO measurement in patients with elevated hematocrits.
Fairbanks and Tefferi (2000) conducted an extensive investigation of normal ranges for
hematocrit and hemoglobin. They analyzed all the published data they could find including
medical textbooks and data from government surveys and hematology laboratories. They found
that when the ranges for anemia are set too low, cases may not be diagnosed and ranges set too
high may result in the misdiagnosis of PV in normal patients. Testing for polycythemia may
mistakenly be undertaken, subsequently diagnosed, and treated in patients with normal values.
Cameskog et aI., (1999) studied 38 patients with apparent polycythemia. They found all patients
had normal RCM measurements with mean hemoglobin levels of 174 gil ± 10 and venous
packed cell volumes (PCV) of 0.53 ±0.03 for males. Fairbanks and Tefferi (2000) concluded
that studies to investigate polycythemia are not usually appropriate for non-iron-deficient adult
Caucasian males with PCV values less than 0.55 (hemoglobin levels less than 180 gIL.) or non
iron-deficient adult Caucasian females with PCV less than 0.50 (hemoglobin concentrations less
than 165 gIL.) (2000). Fairbanks (2000) has reminded clinicians that any adult male with a
hemoglobin of 180gIL and a packed cell volume of 0.54 has approximately a 500 to 1 chance that
he does not have PV. Pearson and Messinezy (1996) recommend at least two blood samples be
drawn before concluding that a patient has an elevated hematocrit. Fluid shifts from fasting

13
states, recumbency, and hydration alter the PCV, blood volume, and hemoglobin level. Blood
drawn in the morning may have vastly different results as opposed to blood drawn from the same
patient in the afternoon (Fairbanks, 2000). See figure 2 algorithm for the evaluation of an
elevated hematocrit.
Pearson et aI., (1995; 1999; 2001) has recommended RCM, measured in relation to body
surface area, for improved accuracy. He proposes RCM measurement in the presence of palpable
splenomegaly when the hematocrit is just below the upper limit of the reference range.
Diagnostic challenges occur when PV exists without splenomegaly or leukocytosis
(Beutler, 2001). Early PV or PV accompanied by iron deficiency may not have an increased
RCM (Tefferi, 1999). Beutler (2001) reveals most cases of PV can be diagnosed without
determining the RCM. Cases of PV with only leukocytosis, thrombocytosis, or splenomegaly
pose a more difficult diagnostic challenge (Beutler, 2001). Lamy et aI., (1997) recommend
RCM measurement for unexplained splenomegaly, thrombocytosis, or hyperleukocytosis and
suggest that the decision to measure RCM not be limited to an elevated hematocrit or
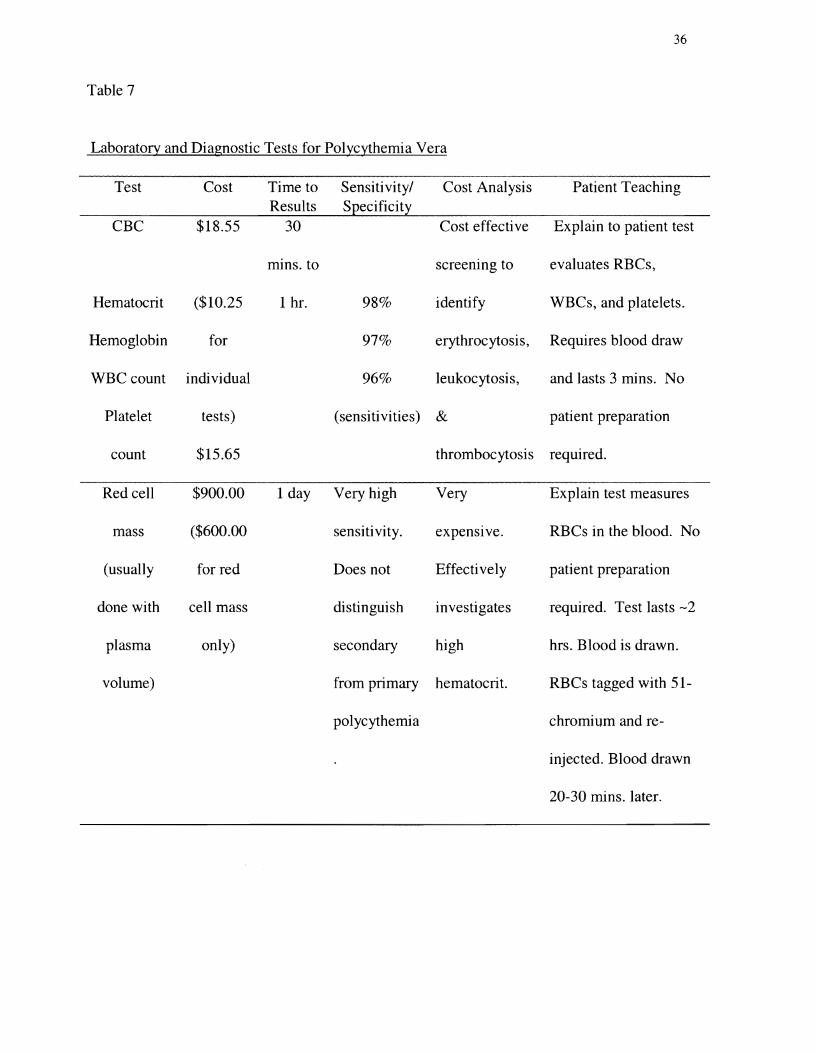
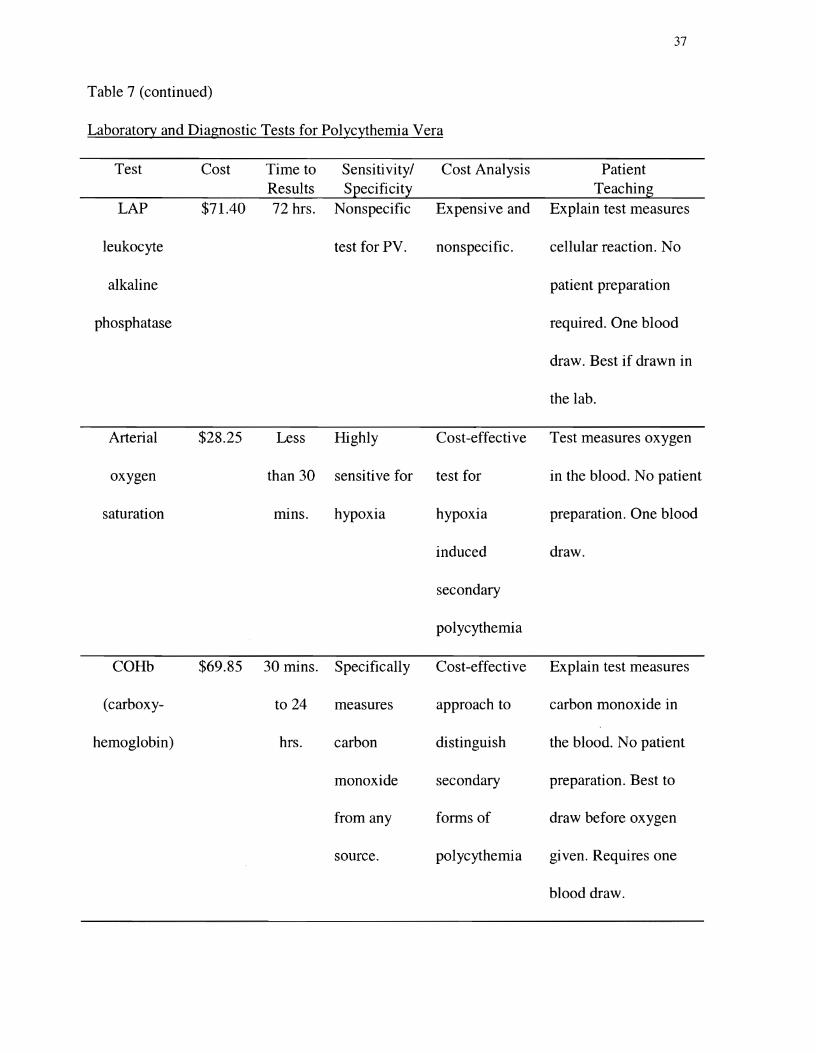
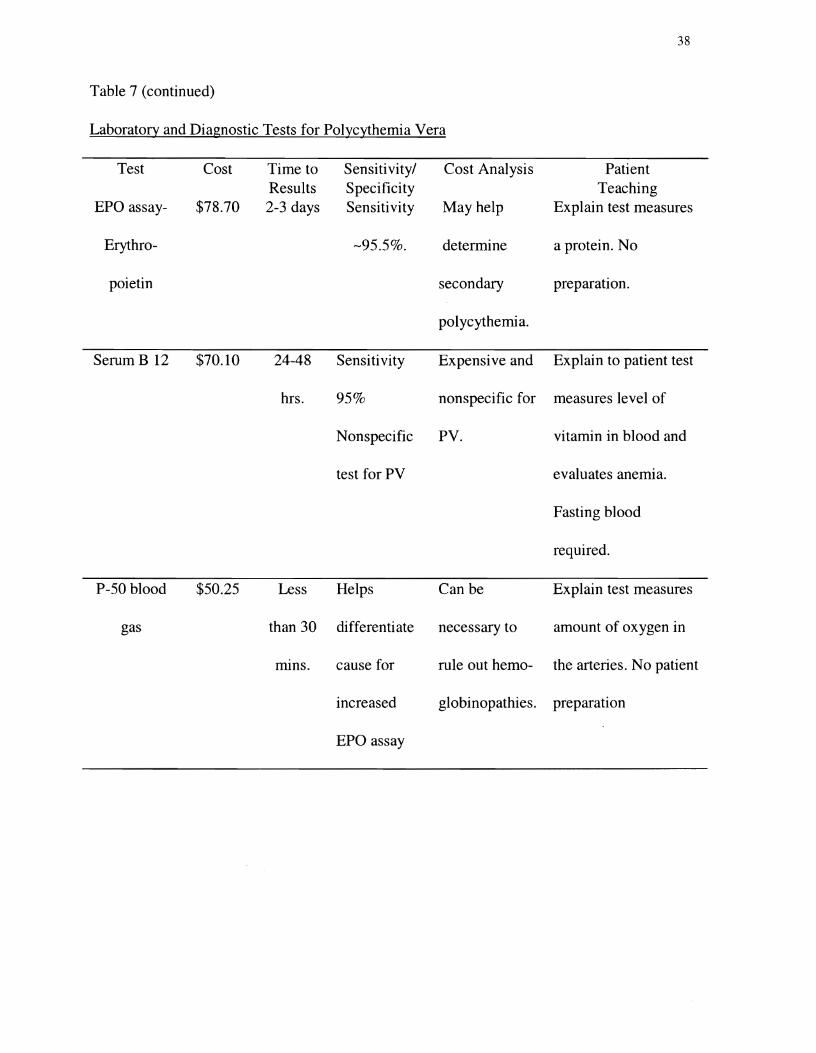
hemoglobin. Table 7 presents tests commonly used to evaluate an elevated hematocrit.
Arterial oxygen saturation can distinguish PV from secondary polycythemia (Berlin,
1975; Beutler, 2001). Arterial oxygen saturation below 92% reveals secondary polycythemia.
Secondary polycythemia due to hypoxia must be evaluated for the underlying cause (Berlin,
1975).
Serum EPO measurement can be useful to differentiate secondary polycythemia from PV
(Napier & Wieczorek, 1981; Silverstein & Tefferi, 2000). A low or absent serum level ofEPO
typically accompanies PV (Adamson & Finch, 1968; Napier & Wieczorek, 1981; de Kleck et aI.,
1981). Secondary polycythemia is usually distinguished by an increased serum EPO (Tefferi,

14
1999). Elevated serum EPa levels due to pathologic production can be caused by cysts in the
liver or kidneys, uterine fibroid tumors, and tumors of the cerebellum, kidneys, or liver. Hypoxic
blood has been has been shown to increase erythropoietin production (Fisher & Langston, 1967).
An elevated serum EPa level without hypoxia raises the suspicion of kidney cysts or tumors
(Taetle, 1998). Tests may be required to confirm the presence of liver or kidney cysts or tumors
in the brain, liver, or kidneys (Talarico, 1998). Sleep studies, abdominal ultrasound,
echocardiogram, liver function tests, and other laboratory tests may be required to investigate
erythrocytosis (Messinezy & Pearson, 1999).
Treatment
Treatment is individualized, based on stage of the disorder, age, sex, clinical
manifestations, and laboratory profile (Hocking, 1995). The risk for thrombohemorrhagic
complications determines whether phlebotomy will be used alone or in combination with
myelosuppressive agents (Tefferi, Solberg, & Silverstein, 2000). Referral to a hematologist for
treatment is recommended. The prevention of thrombotic complications is an important goal of
treatment (Peereboom, 1996). Medications to treat gout, erythromelalgia, and pruritus are often a
necessary part of treatment.
The PVSG data from 1967 to 1987, revealed an increased incidence of thrombosis during
the first 3 years of treatment with phlebotomy alone. They found an increased risk of thrombosis
associated with increasing phlebotomy rates although a causal relationship has not established.
This same subject group had a higher mean survival rate (12.6 years) as opposed to patients
treated more aggressively with medications. The best survival rates have occurred with the use
of phlebotomy alone. The incidence of conversion to acute leukemia was significantly lower
among subjects in the phlebotomy arm of the study. Less than 5% of patients treated with

15
phlebotomy alone experience conversion to acute leukemia (Berk et aI., 1986; Berk, Wasserman,
Fruchtman, & Golclberg, 1995).
Based on the observations by the PVSG all patients with PV should receive phlebotomy
to normalize blood volume (Berk et aI., 1986; Fruchtman & Wasserman, 1995). The goal of
phlebotomy is to keep the hematocrit below 45% in men and below 42% in women (Pearson &
Wetherley-Mein, 1980; Fruchtman & Wasserman, 1995). Pearson and Wetherley (1979) found
that subjects who succumbed to cerebrovascular accidents had venous hematocrits ranging from
0.52 to 0.57. Phlebotomy treatment may initially remove between 250 to 500 cc to reach a
hematocrit between 40 and 45%. Elderly patients and those with cardiovascular disease are less
aggressively phlebotomized. Blood values to determine the frequency of phlebotomy are
monitored every 4 to 8 weeks. Iron deficiency develops with microcytic, hypochromic cells.
Iron supplementation is avoided since it can increase red cell mass and hematocrit (Tefferi et aI.,
2000; Fruchtman & Wasserman, 1995). Phlebotomy does not correct thrombocytosis,
leukocytosis, pruritus, or splenomegaly (Fruchtman & Wasserman, 1995). Elderly patients and
other patients with an increased risk for thrombosis may require additional treatment with
myelosuppressive agents (Fruchtman et aI., 1997).
The myelosuppressive agents frequently used in the United States are interferon alpha,
radioactive phosphorus, and hydroxyurea. Interferon-alpha has been shown to decrease the need
for phlebotomy, and improve splenomegaly, platelet, and hematocrit counts (Taylor et aI., 1996).
Hydroxyurea is well tolerated, easy to administer, and effective in most patients. Radioactive
phosphorus is often used for elderly patients (Fruchtman et aI., 1997). Median survival rate for
patients treated with radioactive phosphorus was 11.8 years in the PVSG (Fruchtman &
Wasserman, 1995).

16
Historically, busulfan has been used for the treatment of PV. Serious side effects have
limited its use and the PVSG no longer recommends alkylating agents due to the risk of cancer
and leukemia (Fruchtman & Wasserman, 1995). The Gruppo Italiano Studio Policitemia (1995)
found a greater than three-fold increase in cancer mortality for patients receiving alkylating
agents or radiophororus. Pipobroman is an alkylating agent used in Europe, but not approved for
use in the United States. A randomized study by Najean and Rain (1997) found no difference in
survival rates among patients given pipobroman or hydroxyurea. Hydroxyurea demonstrated
less effective hematological control in the same study.
Response to myelosuppressive therapy is carefully monitored with frequent
determinations of hematocrit, leukocyte, and platelet counts. Pearson's (1997) in vivo studies
showed elevated packed cell volumes and platelet counts predispose patients to platelet
activation, aggregation, and adhesion. Evaluations for thrombotic complications and progression
to leukemia should be tested routinely (Peereboom, 1996).
The use of aspirin for its antithrombotic properties was investigated by the Gruppo
Italiano Studio Policitemia (1997). The Gruppo Italiano Studio Policitemia conducted a pilot
study to determine the feasibility of a large trial to examine the use of aspirin in patients with PV
(1997). Forty milligrams of aspirin daily inhibited platelet activity and was well tolerated by
study subjects. The PVSG protocol 05 found no reduction in the incidence of thrombosis and an
increase in hemorrhage when aspirin 300 mg and persantine 75 mg were given three times daily
(Tartaglia, Goldberg, Berk, & Wasserman, 1986). Low-dose aspirin may be effective in the
prevention of thrombotic complications. The European Collaboration on Low-dose Aspirin in
Polycythemia Vera (ECLAP) is currently conducting a large-scale randomized trial to evaluate
the benefits and risks of aspirin prophylaxis for thrombotic complications (1997). Tefferi and

17
others are currently using aspirin for the treatment of vasomotor symptoms (2000). Aspirin is
currently indicated for patients with erythromelalgia, transient ischemic attacks, and those with a
history of arterial thrombosis (van Genderen & Troost, 2000).
The control of severe pruritus is important in the total treatment of PV. It may be
alleviated by the use of hydroxyurea or alpha interferon (Taylor et aI., 1996; Tatarsky & Sharon,
1997). Starch baths in lukewarm water and gentle patting instead of rubbing when drying the
skin is recommended (Fruchtman & Wasserman, 1995). Over-the-counter histamine antagonists
such as diphenhydramine or cimetidine have been used to relieve pruritus (Fruchtman &
Wasserman, 1995; Easton & Galbraith, 1978). Antidepressants such as doxepin or paroxetine
given in low doses have also been prescribed for intractable cases of pruritus. Low-dose aspirin
therapy has been recommended for itching and painful burning in the hands and feet (Tefferi,
1998).
Hyperuricemia with acute gout and nephrolithiasis usually improve with
myelosuppressive therapy. Allopurinol may be given to patients treated with phlebotomy alone.
Anti-inflammatory medications or colchicines have been used (Fruchtman & Wasserman, 1995).
Complications
Thrombotic events comprise about one third of the complications found in PV.
Cerebrovascular accidents represent the most frequent thrombotic event followed by myocardial
infarction, deep vein thrombosis, and pulmonary embolism. Bleeding and bruising occurs in
about one fourth of patients (Berk et aI., 1986). Chievitz and Thiede (1962) found hemorrhage
occurred less frequently than thrombosis. The other frequent causes of death include acute
leukemia, neoplasms, hemorrhage, and the "spent" phase with myelofibrosis and myeloid

18
metaplasia (Berk et aI., 1986; Chievitz & Thiede, 1962). Other complications of PV include
peptic ulcer disease and gastrointestinal bleeding (Talarico, 1998; Tefferi, 1998).
Conclusion
Although no cure exists for PV, treatment can prolong lives and enhance th~ quality-of
life for patients affected by PV. The insidious onset may make diagnosis difficult, but PV should
be suspected when any patient presents with an elevated hematocrit. An abnormally elevated
hematocrit accompanied by symptoms such as thrombocytosis, splenomegaly, and leukocytosis,
strongly suggests PV. Prompt referral to a hematologist for bone marrow aspiration and
treatment is recommended.

19
References
Adamson, J.W., Fialkow, P.J., Murphy, S., Prchal, J., & Steinmann, L. (1976).
Polycythemia vera: stem-cell and probable clonal origin of the disease. The New England Journal
of Medicine, 295, 913-916.
Aggeler, P.M., Pollycove, M., Hoag, S., Donald, W., & Lawrence, J. (1960).
Polycythemia vera in childhood. Studies of iron kinetics with Fe59 and blood clotting factors.
Blood, 17, 345-350.
Ansell, J.E. (2001). Cardinal manifestations of hematologic disease, anemias, and related
conditions. In J. Noble, H. L. Greene, W. Levinson, G. A. Modest, C. D. Mulrow, J. E. Scherger,
& M. J. Young (Eds.), Textbook of Primary Care Medicine (3rd ed., pp. 1036-1037). St. Louis:
Mosby.
Asimakopoulos, F.A., Hinshelwood, S., Gilbert, J., Delibrias, C., Gottgens, B., Fearon,
D., & Green, A. R. (1997). The gene encoding hematopoietic cell phosphatase (SlIP-I) is
structurally and transcriptionally intact in polycythemia vera. Oncogene, 14, 1215-1222.
Balcerzak, S.P., & Bromberg, P.A. (1975). Secondary polycythemia. Seminars in
Hematology, 12, 353-382.
Berglund, S., & Zettervall, O. (1992). Incidence of polycythemia vera in a defined
population. European Journal of Haematology, 48, 20-26.
Berk, N.I. (1966). Differential diagnosis of the polycythemias. Seminars in Hematology,
~ 209-213.
Berk, N.I. (1975). Diagnosis and classification of the polycythemias. Seminars in
Hematology, 12,339-351.

20
Berk, P.D. (1997). Epilogue: broader lessons from the study of polycythemia vera.
Seminars in Hematology, (34), 1, 77 - 80.
Berk, P.D., Goldberg, J.D., Donovan, P.B., Fruchtman, S.M., Berlin, N.I., & Wasserman,
L.R. (1986). Therapeutic recommendations in Polycythemia Vera Study Group protocols.
Seminars in Hematology, 23,(2) , 136.
Beutler, E. (2001). Polycythemia vera. In E. Beutler, M.A. Lichtman, B.S. Coller, T.J.
Kipps, & Seligsohn, U. (Eds.), Williams Hematology (6th ed., pp. 689-699). New York:
McGraw-Hill.
Brown, S.M., Gilbert, H.S., Krauss, S., & Wasserman, L. (1971). Spurious (relative)
polycythemia: a nonexistent disease. The American Journal of Medicine, 50, 200-207.
Burge, P.S., Johnson, W.S., & Prankerd, T.A.J. (1975). Morbidity and mortality in
pseudopolycythemia. The Lancet, 1, 1266-1272.
Carneskog, J., Safai-Kutti, S., Suurkula, M., Wadenvik, H., Bake, B., Lindstedt, G., &
Kutti, J. (1999). The red cell mass, plasma erythropoietin and spleen size in apparent
polycythemia. European Journal Haematology, 62, 43-48.
Caldwell, G.G., Kelley, D.B., Heath, C.W., & Zack, M. (1984). Polycythemia vera among
participants of a nuclear weapons test. Journal of the American Medical Association, 252, 662
664.
Chaiter, Y., Brenner, B., Aghai, E., & Tatarsky, I. (1992). High incidence of
myeloproliferative disorders in Ashkenazi Jews in northern Israel. Leukemia and Lymphoma, 7,
251-255.

21
Chien, S., & Gallik, S. (1995). Rheology in normal individuals and polycythemia vera. In
L. R. Wasserman, P. D. Berk, & N. I. Berlin (Eds.), Polycythemia vera and the
myeloproliferative disorders (pp. 114-129). Philadelphia, PA: W. B. Saunders.
Chievitz, E., & Thiede, T. (1962). Complications and causes of death in polycythemia
vera. Acta Medica Scandinavica, 172, 513-523.
Copelan, E.A., & Balcerzak, S.P. (1995). Secondary polycythemia. In L.R. Wasserman,
P.D. Berk & N. I. Berlin (Eds.), Polycythemia vera and the myeloproliferative disorders (pp. 195
221). Philadelphia, PA: W.B. Saunders.
Cotes, P.M., Dore, C.J., Liu Yin, J., Lewis, S., Messinezy, M., Pearson, T., & Reid, C.
(1986). Determination of serum immunoreactive erythropoietin in the investigation of
erythrocytosis. The New England Journal of Medicine, 315, 283-287.
David, A.K. (1995). Hematology. In R.E. Rakel (Ed.), Textbook of Family Practice (5th
ed., pp. 1287-1288). Philadelphia, PA: W.B. Saunders.
de Klerk, G., Rosengarten, P.C.J., Vet, J.W.M., & Goudsmit, R. (1981). Serum
erythropoietin (ESF) titers in polycythemia. Blood, 58, 1171-1174.
Easton, P., & Galbraith, P.R. (1978). Cimetidine treatment of pruritus in polycythemia
vera. The New England Journal of Medicine, 299, 1134.
Eaves, C.J., & Eaves, A.C. (1978). Erythropoietin (EP) dose-response curves for three
classes of erythroid progenitors in normal human marrow and in patients with polycythemia vera.
Blood, 52,1196-1210.
Erslev, A.J., Caro, J., Kansu, E., Miller, 0., & Cobbs, E. (1979). Plasma erythropoietin in
polycythemia. The American Journal of Medicine, 66,243-247.

22
Fairbanks, V. F. (2000). Polycythemia vera: The packed cell volume and the curious
logic of the red cell mass. Hematology, 4, 381-395.
Fairbanks, V.F., & Tefferi, A. (2000). Normal ranges for packed cell volume and
hemoglobin concentration in adults: relevance to 'apparent polycythemia'. European Journal of
Haematology, 65, 285-296.
Fisher, J. W., & Langston, J. W. (1967). The influence of hypoxemia and cobalt on
erythropoietin production in the isolated perfused dog kidney. Blood, 29, 114-125.
Ferri, F.F. (2001). Ferri's Clinical Advisor. (p. 548). St. Louis, MO: Mosby-Year Book.
Fruchtman, S. M., Mack, K., Kaplan, M. E., Peterson, P., Berk, P. D., & Wasserman, L.
R. (1997). From efficacy to safety: A polycythemia vera study group report on hydroxyurea in
patients with polycythemia vera. Seminars in Hematology, 34, 17-23.
Fruchtman, S. M., & Wasserman, L. R. (1995). Therapeutic recommendations for
polycythemia vera. In L. R. Wasserman, P.D. Berk, & N. I. Berlin (Eds.), Polycythemia vera and
the myeloproliferative disorders (pp. 337-349). Philadelphia, PA: W. B. Saunders.
Gruppo Italiano Studio Policitemia. (1995). Polycythemia vera: the natural history of
1213 patients followed for 20 years. Annals of Internal Medicine, 123, 656-664.
Gruppo Italiano Studio Policitemia. (1997). Low-dose aspirin in polycythemia vera: a
pilot study. British Journal of Haematology, 97, 453-456.
Hess, G., Rose, P., Gamm, H., Papadileris, S., Huber, C., & Seliger, B. (1994). Molecular
analysis of the erythropoietin receptor system in patients with polycythaemia vera. British Journal
of Haematology, 88, 794-802.
Hocking, W.G. (1995). Primary and secondary erythrocytosis. In Joseph S. Mazza (Ed.),
Manual of Clinical Hematology (2nd ed., pp.70 - 81). Boston: Little, Brown, and Company.

23
Krajnik, M., & Zylicz, Z. (2001). Understanding pruritus in systemic disease. Journal of
Pain and Symptom Management, 21, 151-168.
Lamy, T., Devillers, A., Bernard, M., Moisan, A., Grulois, I., Drenou, B., Amiot, L.,
Fauchet, R., & Le Prise, P. Y. (1997) . Inapparent polycythemia vera: an unrecognized diagnosis.
The American Journal of Medicine, 102, (1), 14 - 20.
Landaw, S.A. (1996). Deciphering polycythemia. Hospital Practice, (34), 3, 155 - 166.
London, I.M., Shemin, D., West, R., & Rittenberg, D. (1949). Heme synthesis and red
blood cell dynamics in normal humans and in subjects with polycythemia vera, sickle-cell
anemia, and pernicious anemia. Journal of Biological Chemistry, 179, 463-484.
Lopez, A.M. (1996). Erythrocytosis. In H. L. Greene II, R.- M.E. Fincher, W.P. Johnson,
L. Kaufmann, R. Mandel, & G. Morrison (Eds.), Clinical Medicine (2nd ed., pp. 363-365). St.
Louis, MO: Mosby-Year Book, Inc.
Marlow, A.A., & Fairbanks, V.F. (1960). Polycythemia vera in an eleven-year-old girl.
The New England Journal of Medicine, 263, 950-952.
Messinezy, M., & Pearson, T.C. (1999). The classification and diagnostic criteria of the
erythrocytoses (polycythemias). Clinical Laboratory Haematology, 21, 309-316.
Michiels, J.J., Abels, J., Steketee, J., Van Vliet, H., & Vuzevski, V. (1985).
Erythromelalgia caused by platelet-mediated arteriolar inflammation and thrombosis in
thrombocythemia. Annals of Internal Medicine, 102,466-471.
Michiels, J.J., Barbui, T., Finazzi, G., Fuchtman, S. M., Kutti, J., Rain, J. D., Silver, R.
T., Tefferi, A., & Thiele, J. (2000). Diagnosis and treatment of polycythemia vera and possible
future study designs of the PVSG. Leukemia and Lymphoma, 36, 239-254.

24
Michiels, J.J., Kutti, J., Stark, P., Bazzan, M., Gugliotta, L., Marchioli, R.,
Griesshammer, M., van Genderen, P. J. J., Briere, J., Kiladjian, J. J., Barbui, T., Finazzi, G.,
Berlin, N. I., Pearson, T. C., Green, A. C., Fruchtmann, S., Silver, R., Hansmann, E., Wehmeier,
A., Lengfelder, E., Landolfi, R., Kvasnicka, H., Hasselbalch, H., Cervantes, F., Reilly, J.,
Demory, J.-L., Gisslinger, H., Guardiola, Ph., Martyre, M., Le Bousse-Kerdiles, M., & Thiele, J.
(1999). Diagnosis, pathogenesis and treatment of the myeloproliferative disorders essential
thrombocythemia, polycythemia vera and essential megakaryocytic granulocytic metaplasia and
myelofibrosis. The Netherlands Journal of Medicine, 54, 46-62.
Michiels, J.J., & Juvonen, E. (1997). Proposal for revised diagnostic criteria of essential
thrombocytosis and polycythemia vera by the Thrombocythemia Vera Study Group. Seminars in
Thrombosis and Hemostasis, 23, 339-347.
Modan, B. (1995). The epidemiology of polycythemia vera. In L. R. Wasserman, P. D.
Berk, & N. I. Berlin (Eds.), Polycythemia vera and the myeloproliferative disorders (pp. 140
146). Philadelphia, PA: W. B. Saunders.
Modan, B., & Lilienfeld, A. (1965). Polycythemia vera and leukemia - the role of
radiation treatment. Medicine, 44, 305-343.
Moliterno, A.R., Hankins, W.D., & Spivak, J.L. (1998). Impaired expression of the
thrombopoietin receptor by platelets from patients with polycythemia vera. The New England
Journal of Medicine, 338, 572-580.
Murphy, S., Davis, J., Walsh, P., & Gardner, F. (1978). Template bleeding time and
clinical hemorrhage in myelolproliferative disease. Archives of Internal Medicine, 138, 1251
1253.

25
Napier, J.A.F., & Janowska-Wieczorek, A. (1981). Erythropoietin measurements in the
differential diagnosis of polycythaemia. British Journal of Haematology, 48, 393-401.
Najean, Y., & Rain, J.-D. (1997). The very long-term evolution of polycythemia vera: an
analysis of 318 patients initially treated by phlebotomy or 32P between 1969 and 1981. Seminars
in Hematology, 34, 6-16.
Pearson, T.C. (2001). Evaluation of diagnostic criteria in polycythemia vera. Seminars in
Hematology, 38, (Suppl. 2),21-24.
Pearson, T. C. (1997). Hemorheologic considerations in the pathogenesis of vascular
occlusive events in polycythemia vera. Seminars in Thrombosis and Hemostasis, 23, 433-439.
Pearson, T.C., Guthrie, D.L., Simpson, J., Chinn, S., Barosi, G., Ferrant, A., Lewis, S., &
Najean, Y. (1995). Interpretation of measured red cell mass and plasma volume in adults: Expert
panel on radionuclides of the International Counsel for Standardization in Haematology. British
Journal of Haematology, 89,748-756.
Pearson, T. C., & Messinezy, M. (1996). Investigation of patients with polycythemia.
Postgrad Medical Journal, 72,519-524.
Pearson, T.C., & Wetherley-Mein, G. (1979). The course and complications of idiopathic
erythrocytosis. Clinical Laboratory Haematology, 1, 189-196.
Pearson, T. C., & Wetherley-Mein, G. (1980). Plasma and whole blood viscosity in
treated primary polycythemia. Clinical Laboratory Haematology, 2, 73-82.
Peereboom, D.M. (1996). Polycythemia vera. In J. Willis Hurst (Ed.) , Medicine for the
Practicing Physician (4th ed., pp. 862-865). Stamford, CT: Appleton & Lange.

26
Peterson, P., & Ellis, J. T. (1995). The development, morphology and function of normal
bone marrow: a review. In L. R. Wasserman, P. D. Berk, & N. I. Berlin (Eds.), Polycythemia
vera and the myeloproliferative disorders (pp. 1-13). Philadelphia, PA: W. B. Saunders.
Peterson, P., & Wasserman, L. R. (1995). The natural history of polycythemia vera. In L.
R. Wasserman, P. D. Berk, & N. I. Berlin (Eds.), Polycythemia vera and the myeloproliferative
disorders (pp. 14-21). Phildelphia, PA: W. B. Saunders.
Prchal, J.F., Adamson, J.W., Murphy, S., Steinmann, L., & Fialkow, P. (1978).
Polycythemia vera - the in vitro response of normal and abnormal stem cell lines to
erythropoietin. The Journal of Clinical Investigation, 61,1044-1047.
Prchal, J.F., & Axelrad, A.A. (1974). Bone marrow responses in polycythemia vera. The
New England Journal of Medicine, 290, 1382.
Prochazka, A.V., & Markowe, H.L.J. (1986). The epidemiology of polycythemia rubra
vera in England and Wales 1968 - 1982. British Journal of Cancer, 53, 59-64.
Silva, M., Richard, C., Benito, A., Sanz, C., Olalla, I., & Fernandez-Luna, J. (1998).
Expression of Bcl-x in erythroid precursors from patients with polycythemia vera. The New
England Journal of Medicine, 338, 564-571.
Silverstein, M.N. (1974). Postpolycythemia myeloid metaplasia. Archives of Internal
Medicine, 134, 113-115.
Silverstein, M.N., Brown, A.L., & Linman, J.W. (1973). Idiopathic myeloid metaplasia,
its evolution into acute leukemia. Archives of Internal Medicine, 132,709-712.
Silverstein, M.N., & Tefferi, A. (1996). Proliferative disorders of the hematologic system.
In F. Plum & J.C. Bennett (Eds.), Cecil- Textbook of Medicine (20th ed., pp. 920-922).

27
Silverstein, M.N., & Tefferi, A. (2000). Proliferative diseases. In F. Plum & J.C. Bennett
(Eds.), Cecil - Textbook of Medicine (21th ed., pp. 935 - 939).
Smith, J.R., & Landaw, S.A. (1978). Smokers' polycythemia. The New England Journal
of Medicine, 298, 6-10.
Spivak, J.L. (2001). Polycythemia vera and other myeloproliferative diseases. In A.S.
Fauci, E. Braunwald, D. Kasper, S. Hauser, D. Longo, & J. Jameson (Eds.), Harrison's Principles
of Internal Medicine (15th ed., pp. 701-703). New York: McGraw-Hill Company, Inc.
Taetle, R. (1998). Polycythemia. In H.L. Greene IT, W.P. Johnson, & D. Lemcke (Eds.),
Decision Making in Medicine an Algorithmic Approach (2nd ed. , pp. 210-211). S1. Louis, MO:
Mosby, Inc.
Talarico, L.D. (1998). Myeloproliferative disorders: A practical review. Patient Care, 9,
20-33.
Tartaglia, A.P., Goldberg, J.D., Berk, P.D., & Wasserman, L.R. (1986). Adverse effects
of antiaggregating platelet therapy in the treatment of polycythemia vera. Seminars in
Hematology,23, 172-176.
Tatarsky, I., & Sharon, R. (1997). Management of polycythemia vera with hydroxyurea.
Seminars in Hematology,34, 24-28.
Taylor, P.C., Dolan, G. Ng, J-P Paul, B., Collin, R., & Reilly, J.T. (1996). Efficacy of
recombinant interferon-alpha (rIPN-a) in polycythemia vera: a study of 17 patients and an
analysis of published data. British Journal of Haematology,92, 55-59.
Tefferi, A. (1998). The Philadelphia chromosome negative chronic myeloproliferative
disorders: A practical overview. Mayo Clinic Proceedings, 73, 1177-1184.

28
Tefferi, A. (1999). Diagnosing polycythemia vera: A paradigm shift. Mayo Clinic
Proceedings, 74, 159-162.
Tefferi, A., Solberg, L.A., & Silverstein, M.N. (2000). A clinical update in polycythemia
vera and essential thrombocythemia. The American Journal of Medicine, 109, 141-149.
van Genderen, P. J. J., & Troost, M. M. (2000). Polycythaemia vera and essential
thrombocythaemia in the elderly. Drugs and Aging, 2, 107-119.
Waldmann, T. A., Rosse, W., & Swarm, R. (1968). The erythropoiesis-stimulating
factors produced by tumors. Annals of the New York Academy of Sciences, 149,509-515.
Wasserman, L.R. (1954). Polycythemia vera - its course and treatment: relation to�
myeloid metaplasia and leukemia. Bulletin of the New York Academy of Medicine, 30, 343-375.�
Watts, E.J., & Lewis, S.M. (1983). Spurious polycythemia - a study of 35 patients.�
Scandinavian Journal of Haematology, 31, 241-247.
Weinberg, R.S., Worsley, A., Gilbert, H., Cuttner, J., Berk, P., & Alter, B. (1989).
Comparison of erythroid progenitor cell growth in vitro in polycythemia vera and chronic
myelogenous leukemia: only polycythemia vera has endogenous colonies. Leukemia Research,
~331-338.
Weinreb, N. J. (1995). Relative polycythemia. In L. R. Wasserman, P. D. Berk, & N. I.
Berlin (Eds.), Polycythemia vera and the myeloproliferative disorders (pp. 226-258).
Philadelphia, PA: W. B. Saunders.
Weinreb, N.J., & Shih, C.F. (1975). Spurious polycythemia. Seminars in Hematology, 12,
397-407.

29
Zanjani, E.D., Lutton, J.D., Hoffman, R., & Wasserman, L. (1977). Erythroid colony
formation by polycythemia vera bone marrow in vitro. The Journal of Clinical Investigation, 59,
841-848.

30
Table 1
Occurrence of Signs and Symptoms in Patients with Polycythemia Vera
Sign/Symptom
Ruddy cyanosis
Palpable splenomegaly
Palpable hepatomegaly
Generalized pruritus
Headache, dizziness, epigastric distress, fatigue
Thrombotic events
Hemorrhagic events including gastrointestinal bleeding
Increased blood pressure
Thrombocytosis
Leukocytosis
Hyperuricemia
Elevated LAP
Mild basophilia
Percentage
- 700/0
- 70-75%
- 40-50%
- 40-50%
-30%
-20%
- 15-350/0
-30%
- 25-500/0
-60%
-40%
-70%
-6.0%
Note. LAP =leukocyte alkaline phosphatase. From (Silverstein & Tefferi, 1996; Hocking,
1995; Tefferi, 1998)

31
Table 2
Differential Diagnosis of Polycythemia Vera
Type of Polycythemia
Spurious/relative/stress polycythemia
Dehydration
Secondary polycythemia
Chronic pulmonary disease
Right to left cardiovascular shunts
High altitude
Erythropoietin-producing neoplasms
Sleep apnea syndrome
Benign renal lesions
Carbon monoxide intoxication
High oxygen affinity hemoglobinopathy
High serum level of carboxyhemoglobin: smoking
Polycythemia Vera
32
Table 3
Laboratory Differentiation of Polycythemia
Test Relative Polycythemia Secondary Polycythemia Polycythemia Vera
Red cell mass N I I
Leukocyte count N N I
Platelet count N N I
Arterial oxygen N NorD N saturation
Erythropoietin level N I NorD
Leukocyte alkaline N N I phosphatase
Serum vitamin B12 N N I
Bone marrow N Erythroid hyperplasia Hypercellular (panmyelosis)
Note. N, normal; D, decreased; I, increased. The differences listed are not present in all patients.
From (Talarico, 1998; Beutler, 2001; Ansell, 2001)

33
Table 4
PVSG Criteria for the Diagnosis of Polycythemia Vera
AI. Increased RBC mass B1. Thrombocytosis:
Platelet count !A 400,000/yl Male: ~ 36 ml/kg
Female: ~ 32 ml/kg B2. Leukocytosis: !A 12,000/yl (no fever or
A2. Normal arterial infection)
oxygen saturation (~92%)
B3. Increased leukocyte alkaline phosphatase A3. Splenomegaly
(LAP !A 100)
B4. Increased serum BD al binders BD a: (!A 900 pg/ml)
Unbound B D a binding capacity: (!A2200 pg/ml)
Note. PVSG =Polycythemia Vera Study Group; RBC =red blood cells; ml =milliliters; kg =
kilograms; yL =microliter; pg =picogram. Diagnosis of polycythemia vera virtually certain in
the presence of Al + A2 + A3 or Al + A2 + any two from category B. From "Therapeutic
Recommendations in Polycythemia Vera Based on Polycythemia Vera Study Group Protocols,"
by P.D. Berk, J.D. Goldberg, P.B. Donovan, S.M. Fruchtman, N.I. Berlin, and L.R. Wasserman,
1986, Seminars in Hematology, 23, p. 134. Copyright 1997 by W.B. Saunders. Reprinted with
permission.

34
Table 5
The Rotterdam Criteria of Polycythemia Vera Proposed by the Thrombocythemia Vera Study Group
(TVSG)
Al Raised red cell mass: B I Thrombocytosis
male> 36 mL/kg Platelet count> 400 x 109/L
female> 32 mL/kg
A2 Absence of any cause of secondary B2 Granulocytes> 10 x 109/L and/or
erythrocytosis by clinical and laboratory raised neutrophil alkaline phosphatase
investigations score of >100 in the absence of fever
or infection
A3 Histopathology of bone marrow biopsy B3 Splenomegaly on palpation or isotope/
increase of ultrasound scan
a.� Cellularity, panmyelosis B4 Erythroid colony formation
b.� Enlarged megakaryocytes with in absence of Epo: spontaneous EEC
Hyperploid nuclei; clusters of
megakaryocytes
c.� reticulin fibers (optional)
A I+A2+A3 is consistent with early stage PV (so-called "idiopathic erythrocyt<?sis")
AI+A2+A3+ anyone from category B establishes overt PV
A3+B I is consistent with essential thrombocythemia
A3+B3 and/or B4 is consistent with a primary myeloproliferative disorder
Note. Epo =erythropoietin, ml =milliliters, kg =kilogram, EEC =endogenous erythroid colony.
From "Proposal for Revised Diagnostic Criteria of Essential Thrombocythemia and Polycythemia
Vera by the Thrombocythemia Vera Study Group," by J. J. Michiels and E. Juvonen, 1997, Seminars
in Thrombosis and Hemostasis, 23, p. 341. Permission for reprint granted. Thieme Medical
Publishers, Inc.

35
Table 6
Proposed Modified Criteria for the Diagnosis of Polycythemia Vera
Al Raised red cell mass B1 Thrombocytosis
(>25% above mean normal predicted value) (platelet count >400xl09/1)
A2 Absence of cause of secondary erythrocytosis B2 Neutrophil leucocytosis
A3 Palpable splenomegaly (neutrophil count>1Ox109/1)
A4 Clonality marker 3/4 acquired abnormal B3 Splenomegaly demonstrated
marrow karyotype� on isotope/ultrasound scanning
B4 Characteristic BFU-E growth
or reduced serum erythropoietin
Al+A2+A3 or A4 establishes PV
Al+A2+ two of B establishes PV
Note. BFU-E =burst forming units - erythroid; PV =polycythemia vera. From "The
Classification and Diagnostic Criteria of the Erythrocytoses (Polycythaemias)" by T. C.
Pearson and M. Messinezy, 1999, Clinical Laboratory Haematology, 21, p. 313. Copyright
1999, Blackwell Science Limited. Permission for reprint granted.
36
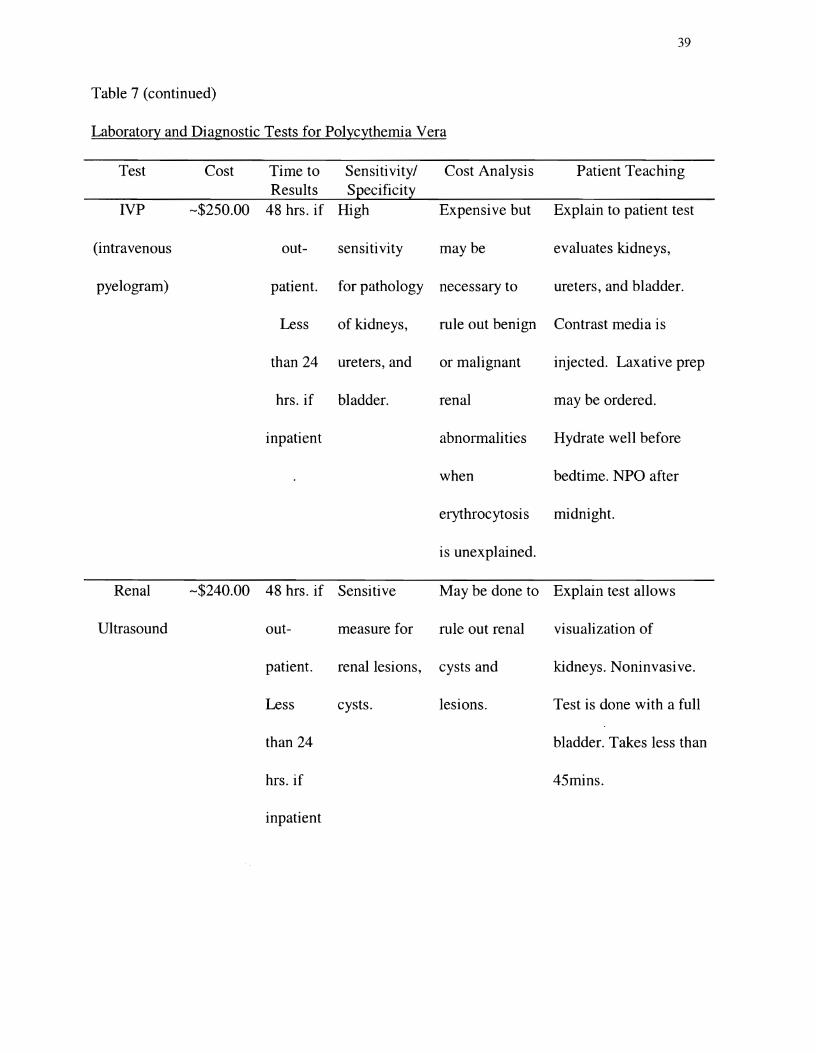
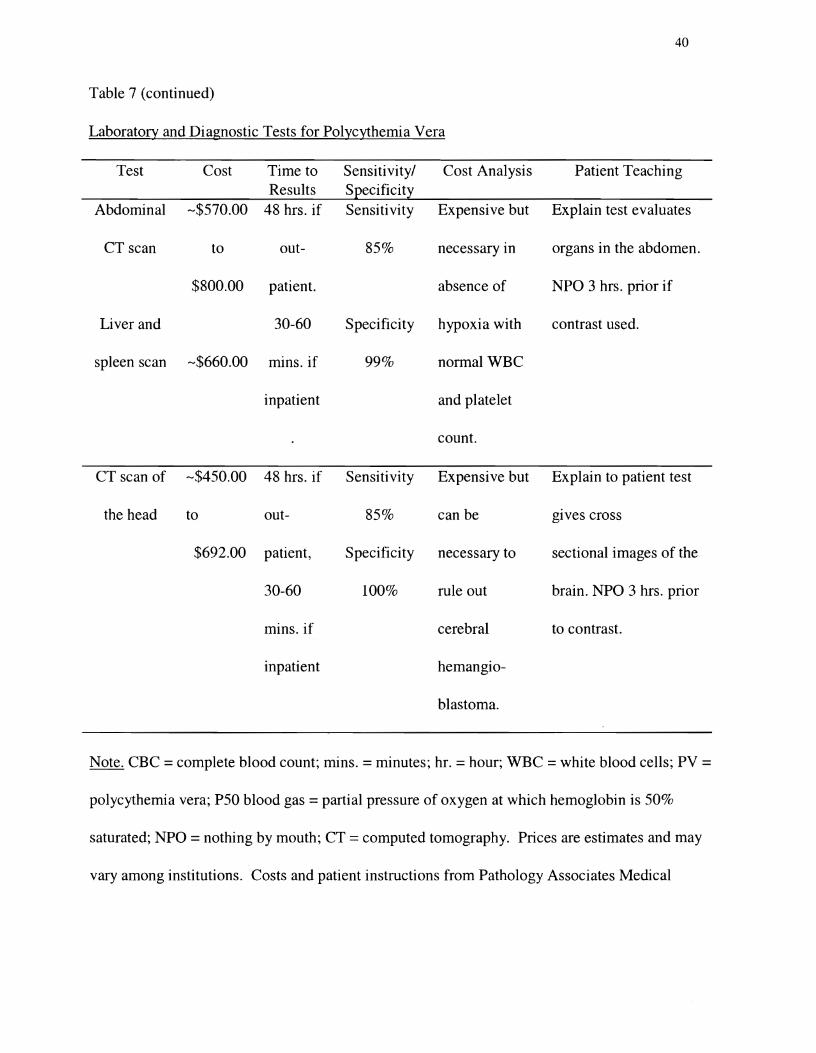
Table 7
Laboratory and Diagnostic Tests for Polycythemia Vera
Test Cost Time to Sensitivity/ Cost Analysis Patient Teaching Results Specificity
CBC $18.55 30 Cost effective Explain to patient test
mins. to screening to evaluates RBCs,
Hematocrit ($10.25 1 hr. 98% identify WBCs, and platelets.
Hemoglobin for 97% erythrocytosis, Requires blood draw
WBC count individual 96% leukocytosis, and lasts 3 mins. No
Platelet tests) (sensitivities) & patient preparation
count $15.65 thrombocytosis required.
Red cell $900.00 1 day Very high Very Explain test measures
mass ($600.00 sensitivity. expensIve. RBCs in the blood. No
(usually for red Does not Effectively patient preparation
done with cell mass distinguish investigates required. Test lasts -2
plasma only) secondary high hrs. Blood is drawn.
volume) from primary hematocrit. RBCs tagged with 51
polycythemia chromium and re
injected. Blood drawn
20-30 mins. later.
37
Table 7 (continued)�
Laboratory and Diagnostic Tests for Polycythemia Vera�
Test Cost Time to Sensitivity/ Cost Analysis Patient Results Specificity Teaching
LAP $71.40 72 hrs. Nonspecific Expensive and Explain test measures
leukocyte test for PV. nonspecific. cellular reaction. No
alkaline patient preparation
phosphatase required. One blood
draw. Best if drawn in
the lab.
Arterial $28.25 Less Highly Cost-effective Test measures oxygen
oxygen than 30 sensitive for test for in the blood. No patient
saturation mIns. hypoxia hypoxia preparation. One blood
induced draw.
secondary
polycythemia
COHb $69.85 30 mins. Specifically Cost-effective Explain test measures
(carboxy to 24 measures approach to carbon monoxide in
hemoglobin) hrs. carbon distinguish the blood. No patient
monoxide secondary preparation. Best to
from any forms of draw before oxygen
source. polycythemia given. Requires one
blood draw.
38
Table 7 (continued)�
Laboratory and Diagnostic Tests for Polycythemia Vera�
Test Cost Time to Sensitivity/ Cost Analysis Patient Results Specificity Teaching
EPO assay $78.70 2-3 days Sensitivity May help Explain test measures
Erythro -95.5%. determine a protein. No
poietin secondary preparation.
polycythemia.
Serum B 12 $70.10 24-48 Sensitivity Expensive and Explain to patient test
hrs. 950/0 nonspecific for measures level of
Nonspecific PV. vitamin in blood and
test for PV evaluates anemia.
Fasting blood
required.
P-50 blood $50.25 Less Helps Can be Explain test measures
gas than 30 differentiate necessary to amount of oxygen in
nuns. cause for rule out hemo the arteries. No patient
increased globinopathies. preparation
EPO assay
39
Table 7 (continued)�
Laboratory and Diagnostic Tests for Polycythemia Vera�
Test Cost Time to Sensitivity/ Cost Analysis Patient Teaching Results Specificity
IVP -$250.00 48 hrs. if High Expensive but Explain to patient test
(intravenous out- sensitivity maybe evaluates kidneys,
pyelogram) patient. for pathology necessary to ureters, and bladder.
Less of kidneys, rule out benign Contrast media is
than 24 ureters, and or malignant injected. Laxative prep
hrs. if bladder. renal may be ordered.
inpatient abnormalities Hydrate well before
when bedtime. NPO after
erythrocytosis midnight.
is unexplained.
Renal -$240.00 48 hrs. if Sensitive May be done to Explain test allows
Ultrasound out- measure for rule out renal visualization of
patient. renal lesions, cysts and kidneys. Noninvasive.
Less cysts. lesions. Test is done with a full
than 24 bladder. Takes less than
hrs. if 45mins.
inpatient
40
Table 7 (continued)�
Laboratory and Diagnostic Tests for Polycythemia Vera�
Test Cost Time to Sensitivity/ Cost Analysis Patient Teaching Results Specificity
Abdominal -$570.00 48 hrs. if Sensitivity Expensive but Explain test evaluates
CT scan to out 85% necessary in organs in the abdomen.
$800.00 patient. absence of NPO 3 hrs. prior if
Liver and 30-60 Specificity hypoxia with contrast used.
spleen scan -$660.00 mins. if 99% normal WBC
inpatient and platelet
count.
CT scan of -$450.00 48 hrs. if Sensitivity Expensive but Explain to patient test
the head to out- 850/0 can be gIves cross
$692.00 patient, Specificity necessary to sectional images of the
30-60 100% rule out brain. NPO 3 hrs. prior
mins. if cerebral to contrast.
inpatient hemangio
blastoma.
Note. CBC =complete blood count; mins. =minutes; hr. =hour; WBC =white blood cells; PV =
polycythemia vera; P50 blood gas = partial pressure of oxygen at which hemoglobin is 50%
saturated; NPO =nothing by mouth; CT =computed tomography. Prices are estimates and may
vary among institutions. Costs and patient instructions from Pathology Associates Medical

41
Laboratory, Inland Imaging, Deaconess Hospital, and Holy Family Hospital, Spokane,
Washington.

42
Table 8�
Patient Education Resources�
National Organization for Rare Disorders, Inc. (NORD)�
Post Office Box 8923�
New Fairfield, CT
Web-Site: http://www.raredisease.orgl�
06812-8923�
Myeloproliferative Disease Research Center, Inc.�
115 East 72nd Street�
New York, NY 10021�
Web Site: http://www.acor.orgl�
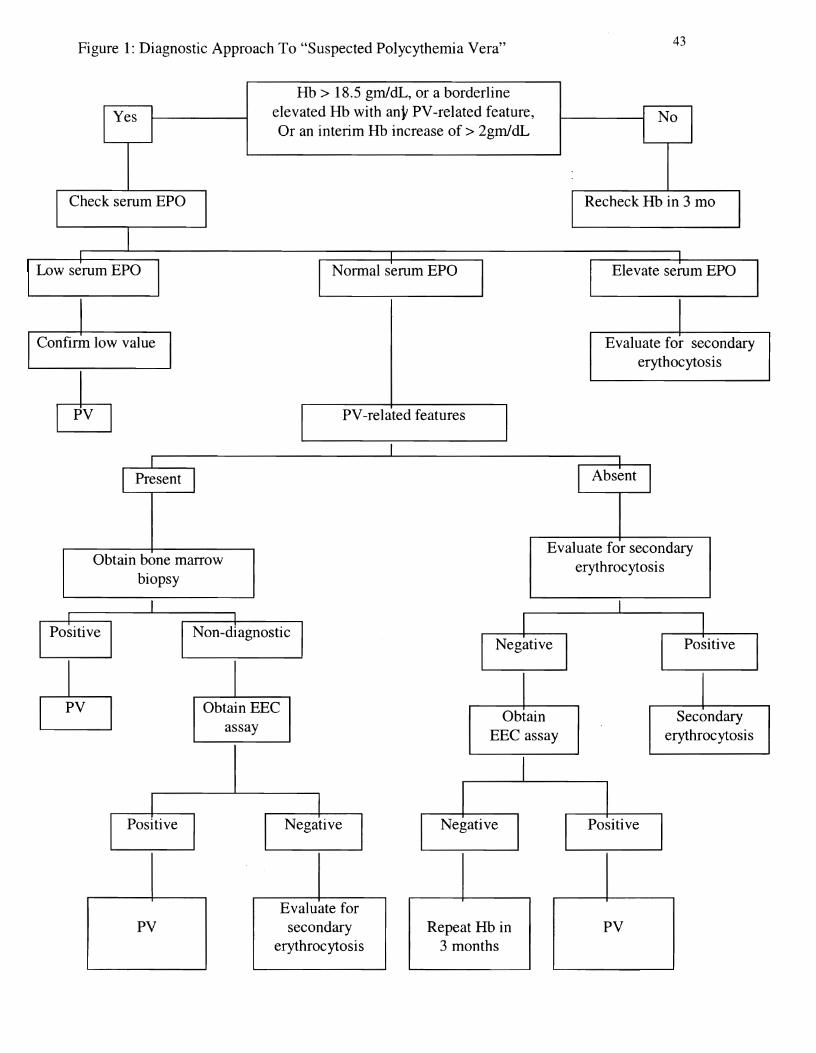
Figure 1: Diagnostic Approach To "Suspected Polycythemia Vera" 43
Hb > 18.5 gm/dL, or a borderline elevated Hb with any PV-related feature, Or an interim Hb increase of > 2gm/dL
Check serum EPO Recheck Hb in 3 mo
Elevate serum EPO
Confirm low value Evaluate for secondary erythocytosis
PV-related features
Obtain bone marrow biopsy
Evaluate for secondary erythrocytosis
Obtain EEC Obtain Secondary
assay EEe assay erythrocytosis
Evaluate for PV secondary Repeat Hb in PV
erythrocytosis 3 months
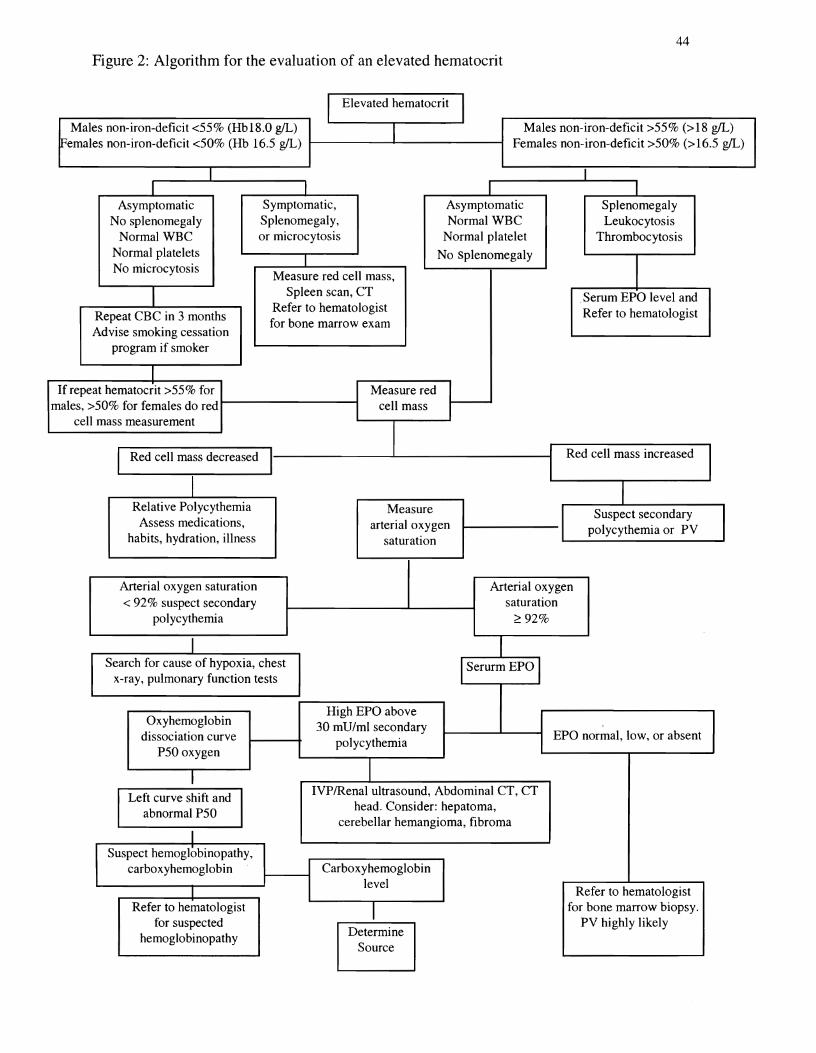
44 Figure 2: Algorithm for the evaluation of an elevated hematocrit
I Elevated hematocrit
Males non-iron-deficit <55% (HbI8.0 gIL) Males non-iron-deficit >55% (> 18 gIL)I[Females non-iron-deficit <50% (Hb 16.5 gIL) Females non-iron-deficit >50% (>16.5 gIL)
I I I I
Asymptomatic Symptomatic, Asymptomatic Splenomegaly No splenomegaly Splenomegaly, Normal WBC Leukocytosis
Normal WBC or microcytosis Normal platelet Thrombocytosis Normal platelets No splenomegaly I INo microcytosis
Measure red cell mass, Spleen scan, CTI Serum EPO level and
Refer to hematologist Refer to hematologist Repeat CBC in 3 months for bone marrow exam
Advise smoking cessation� program if smoker�
I If repeat hematocrit >55% for Measure red
males, >50% for females do red t------------t cell mass cell mass measurement
I I Red cell mass increased I Red cell mass decreased I I
I IRelative Polycythemia Measure Suspect secondary
Assess medications, arterial oxygen polycythemia or PVI Ihabits, hydration, illness saturatIon
Arterial oxygen saturation Arterial oxygen� < 92% suspect secondary saturation�
polycythemia ~92%
I Search for cause of hypoxia, chest�
x-ray, pulmonary function tests�
High EPO above Oxyhemoglobin 30 mU/ml secondary
dissociation curve EPO normal, low, or absent polycythemia
P50 oxygen
IVPlRenal ultrasound, Abdominal CT, CTLeft curve shift and head. Consider: hepatoma, abnormal P50
cerebellar hemangioma, fibroma I
Suspect hemoglobinopathy,� carboxyhemoglobin Carboxyhemoglobin�
level�I Refer to hematologist
Refer to hematologist for bone marrow biopsy. Ifor suspected PV highly likely Determinehemoglobinopathy
Source

45
Figure Caption
Figure 1 Diagnostic Approach to "Suspected Polycythemia Vera." From "Diagnosing
Polycythemia Vera: A Paradigm Shift," by A. Tefferi, 1999, Mayo Clinic Proceedings, 74, p.
161. Copyright 1999 by Mayo Foundation for Medical Education and Research. Permission to
reprint granted. Of note, hemoglobin (Hb) level is for male Caucasians. Hb value for female
Caucasians is 17 gmJdL; for male African-Americans, 17.5 gmJdL; and for female
African=Americans, 16.7 gmJdL. EEC = endogenous erythroid colony; EPO = erythropoietin;
PV = polycythemia vera.
Figure 2 . Algorithm for the evaluation of an elevated hematocrit. Diagnostic approaches to
elevated hematocrits are based on signs, symptoms, and laboratory findings. WBC = white blood
cell; CBC = complete blood count; EPO = erythropoietin; CT = computed tomography; mD =
milliunit; ml = milliliter; IVP = intravenous pyelogram; PV = polycythemia vera; P50 = partial
pressure of oxygen at which hemoglobin is 50% saturated.