thrombotic microangiopathies: towards a pathophysiology-based
TRANSCRIPT

Cardiovascular & Haematological Disorders-Drug Targets, 2009, 9, 000-000 1
1871-529X/09 $55.00+.00 © 2009 Bentham Science Publishers Ltd.
Thrombotic Microangiopathies: Towards a Pathophysiology-Based Classification
Paul Coppo1,*
and Agnès Veyradier2
1Service d’Hématologie et de Thérapie Cellulaire, Hôpital Saint-Antoine, AP-HP, UPMC Univ Paris 6, Paris, France;
2Service d’Hématologie Biologique, Hôpital Antoine Béclère, AP-HP, Université Paris XI, Clamart, France
Abstract: Thrombotic microangiopathies (TMA) encompass various diseases characterized by a microangiopathic hemo-
lytic anemia, platelet clumping, and organ failure of variable severity. Thrombotic thrombocytopenic purpura (TTP) is a
particularly severe form of TMA characterized by systemic organ failure which results from a severe defect in
ADAMTS13, a plasma enzyme specifically involved in the cleavage of highly hemostatic unusually large (UL) von
Willebrand factor (VWF) multimers into smaller and less adhesive VWF forms. Failure to degrade these UL-VWF mul-
timers leads to excessive platelet aggregates and capillary occlusion. ADAMTS13 deficiency results from bi-allelic muta-
tions in hereditary TTP, whereas in acquired forms it results from autoantibodies that alter the protein function. Patients
with acquired idiopathic TTP have a trend to develop autoimmunity, since a clinical context of autoimmunity may be
found in 30p. cent of cases. Moreover, the remarkable efficiency of monoclonal antibodies directed against CD20 antigen
of B lymphocytes in refractory or chronic relapsing forms provides an additional indirect argument to consider acquired
TTP as an autoimmune disease.
Hemolytic uremic syndrome (HUS) is characterized prominently by a renal failure. In most cases, HUS is caused by en-
tero-hemorrhagic Escherichia coli (diarrhea-positive HUS). Diarrhea-negative HUS, termed atypical HUS, was associated
with a dysfunction in complement pathway involving mutations in factor H, factor I, CD46/MCP, factor B and C3 com-
ponents.
The major improvement in our understanding of TMA pathophysiology allows now a more accurate molecular classifica-
tion of TMA syndromes, which opens fascinating perspectives of targeted therapies in the forthcoming years.
Key Words : Thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, thrombotic microangiopathies, thrombocy-
topenia, ADAMTS13, autoimmune disease, complement.
INTRODUCTION
Thrombotic microangiopathies (TMA) encompass a het-
erogeneous group of disorders characterized by the associa-
tion of a microangiopathic hemolytic anemia (Fig. 1A) with
peripheral thrombocytopenia and organ injury of variable
severity, that result from microthrombi in capillaries and
arterioles (Fig. 1B). These diseases can affect people of all
classes of age, although some forms are more frequently
observed in children, while others are more likely encoun-
tered in adult population. Distinct subsets of TMA can be
distinguished depending on clinical presentation and physio-
pathological mechanisms. In 1924, Moschcowitz described
thrombotic thrombocytopenic purpura (TTP) as an acute
febrile pleiochromic anemia with hyaline thrombi in the ter-
minal arterioles and capillaries of the brain, heart, pancreas,
spleen, kidney and adrenal gland [1]. In hemolytic uremic
syndrome (HUS), severe acute renal failure with bilateral
necrosis of the renal cortex is the prominent feature, and
results from platelet-fibrin thrombi occluding predominantly
the renal circulation [2]. A TMA syndrome can also be
*Address correspondence to this author at the Service d’Hématologie et de
Thérapie Cellulaire, Hôpital Saint-Antoine, 184 rue du Fbg St Antoine
75012 Paris, France; Tel: 00*33 1 49 28 26 21; Fax: 00*33 1 49 28 32 00;
E-mail: [email protected]
observed in association with pregnancy, cancer and chemo-
therapy, human immunodeficiency (HIV) infection, malig-
nant hypertension, or following hematopoietic stem cell
transplantation.
Advances in recent years have delineated the molecular
mechanisms of some TMA syndromes, including TTP, some
atypical HUS and HELLP syndrome. These studies have
clearly revealed that TMA syndromes encompass several
distinct molecular defects, although the cause of TMA might
not be immediately apparent from the clinical presentation.
Concerning nomenclature and syndromic definitions, we use
here distinctively the terms TTP and HUS to identify TMA
syndromes fulfilling typical clinical and biological presenta-
tions of these respective entities. When the underlying cause
of disease is uncertain, the term TMA is preferred. This
work, based on the experience of the French group, aims to
present the most recent physiopathological and therapeutical
data in the field of TTP, HUS, and other TMA syndromes.
THE FRENCH REFERENCE CENTRE FOR THE
MANAGEMENT OF THROMBOTIC MICROAN-
GIOPATHIES
The French network on Thrombotic Microangiopathies is
a multidisciplinary collaborative group involving hematolo-
gists, ressuscitators, nephrologists, immunopathologists and

2 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
biologists from more than 30 national sites, which objective
is to coordinate the management and the study of TMA at the
national level. For this aim, the French Network set up a
prospective registry which currently involves more than 650
patients exhaustively studied between 2000 and 2008. This
approach allowed improving considerably our experience in
the field of these rare diseases, which now allows drawing
consensual diagnostic and therapeutical guidelines. In 2006,
our Network was designated by the French Ministry of
Health as the official national reference center for these rare
diseases, which helped strengthening the organization at the
national level and improving the distributed information
among health actors, patients and their families.
THROMBOTIC THROMBOCYTOPENIC PURPURA
Pathophysiology
Infections and Endothelial Activation
Despite their sharp heterogeneity, TMA share some
common pathophysiological mechanisms. Particularly, endo-
thelium damage and activation may be a general phenome-
non involved in the initiation of a TMA episode [3] [4]. Fac-
tors involved in this early process may include infections,
medications, immune complexes, cancer cells, or alloreactiv-
ity. The association between TTP and infections as triggering
factors is well recognized for many years [5]. Various patho-
gens (bacterials, viruses, fungal agents or parasites) could be
evidenced at the acute phase of TTP, or within the preceding
days. TTP was also reported after administration of vaccinal
antigens. Multiple mechanisms may be involved in endothe-
lial activation by microbial-derived antigens. In vitro studies
provided evidence that bacterial structures such as lipopoly-
saccharide (LPS) act synergistically with various mediators
of inflammation to activate endothelium. These include in-
terleukin (IL)-1, IL-6, interferon- , tumor necrosis factor
(TNF)- and Fas-ligand [6] [7]. As a consequence, endothe-
lial cells acquire a proaggregant phenotype by releasing
highly adhesive von Willebrand factor (VWF) multimers and
by expressing adhesion molecules at their surface. On the
opposite, they decrease the release of prostaglandin-I2, a
strong endogenous antiplatelet agent. In patients with deter-
mined risk factors for TTP, these features lead to a persistent
platelet aggregation with thrombi formation and systemic
microvessels occlusion.
Role of Von Willebrand Factor and ADAMTS13 Defi-
ciency
VWF is a multimeric protein involved in the initiation of
platelet clumping. It is synthesized and released by endothe-
lial cells and stored in cytoplasmic organelles called Weibel-
Palade granules. The basal structure of VWF is a 200 to 300
kDa monomer. Those monomers are linked by disulfide
bonds and form multimers of 500 to 20-30000 kDa with a
globular conformation which diminishes VWF-platelet inter-
action. In high shear stress conditions, however, those high
molecular weight VWF multimers become unfolded and
display many binding sites for ligands, enhancing their he-
mostatic activity [8].
The link between VWF and TTP pathophysiology was
provided by the seminal work of Moake [9], who found that
patients with chronic relapsing TTP displayed large amounts
of circulating high molecular weight VWF at the acute phase
of the disease and during remission. Since these unusually
large VWF multimers are absent in normal plasma, Moake
hypothesized that a yet undiscovered plasma protease was
involved in the cleavage of hyper-adhesive VWF multimers.
This protein was purified in 1996 by Tsai and Furlan inde-
pendently [10] [11], and cloned in 2001 [12]. It is a specific
zinc metalloproteinase called ADAMTS13 (acronym for A
Disintegrin And Metalloproteinase with ThromboSpondin-1
motifs), that specifically cleaves unfolded, high molecular
weight VWF multimers between tyrosine 842 and methio-
nine 843 residues of A2 domain in high shear stress condi-
tion (Fig. 2). As a consequence, a dysfunction of ADAMTS13
leads to a persistent VWF-dependent platelet accumulation,
eventually causing microvascular thrombosis and TTP (Fig.
3).
The ADAMTS13 gene contains 29 exons spanning ap-
proximately 37 kb on chromosome 9q34. It encodes a 4.7-kb
transcript that is expressed primarily in the stellate cells of
the liver (Ito cells) and a 2.4-kb transcript in multiple tissues
including placenta, skeletal muscle and certain tumor cell
lines. Plasma ADAMTS13 concentration in about 1 g/ml
and its evaluated plasmatic half-life is 3 days [13]. Until
Fig. (1). (A) blood smear disclosing fragmented red cells or
schistocytes. Red cells undergo fragmentation in microvasculature,
which calibre is severely reduced by platelet thrombi. Schistocytes
may undergo multiple fragmentations. (B) Typical histopathologi-
cal features of thrombotic microangiopathy (TMA) on a renal
biopsy. Multiple thrombi are observed in glomerular capillaries and
in arterioles (red staining). Thrombi may predominantly be made of
von Willebrand factor (TTP), or either fibrin (HUS).

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 3
now, ADAMTS13 remains the only enzyme involved in the
cleavage of VWF multimers. However, it is not excluded
that other proteins may act in concert with ADAMTS13 to
process VWF multimers. Indeed, thrombospondin-1 (TSP-1)
is an enzyme with reductase activity towards VWF disulfide
bonds, which may facilitate ADAMTS13 access to VWF
cleavage sites [14]. By regulating VWF multimers size,
ADAMTS13 was also reported to have a role in the regula-
tion of leukocytes rolling and adhesion on endothelial cells
and in neutrophils extravasation during inflammation [15]. In
addition, although not yet observed in humans, complete
deficiency of ADAMTS13 in mice was reported to be
prothrombotic, even in the absence of findings consistent
with TTP [16].
In normal healthy individuals, ADAMTS13 activity
ranges between 50 and 150 p. cent. ADAMTS13 activity is
often mildly decreased (20 to 40 p. cent of activity) in vari-
ous contexts such as disseminated intravascular coagulation,
liver cirrhosis, severe sepsis, idiopathic thrombocytopenic
purpura, or systemic lupus erythematosus (not associated
with TMA) [17] [18]. A low ADAMTS13 activity was also
reported in particular physiological conditions, such as eld-
erly, neonates, during pregnancy, or after a surgical proce-
dure [19] [18]. The significance of this result is still unclear;
it may result from a transient disturbance in protein synthe-
sis, and/or consumption in relation with an increase in circu-
lating VWF multimers concentrations. These results are in
sharp contrast to the extremely low levels (less than 5 p. cent
of normal values) of ADAMTS13 activity observed in 70 to
more than 90 p. cent of patients with TTP [20] [21] [22].
Moreover, ADAMTS13 activity was found normal or at least
detectable in most HUS cases [20] [23], or in other TMA
syndromes including hematopoietic stem cell transplanta-
tion-associated TMA, HELLP syndrome, or catastrophic
antiphospholipid syndrome.
Mecjmhanisms of ADAMTS13 Deficiency in TTP
The severely decreased ADAMTS13 activity observed in
TTP may result from biallelic mutations of ADAMTS13
gene (5 to 10 p. 100 of cases), which mainly involve pediat-
ric cases, or from autoantibodies directed to ADAMTS13 (90
to 95 p. 100 of cases), corresponding mostly to adult forms
[13].
ADAMTS13 Deficiency in Congenital TTP
Congenital TTP, previously known as Upshaw-Schulman
syndrome, is an autosomal recessive disease resulting from
biallelic mutations of ADAMTS13 gene. Until now, more
than 70 candidate mutations were reported, which involve
amino-acid substitutions (60 p. cent of cases), non-sense
mutations, or frameshift mutations (20 p. cent both). Half
mutations occur in the catalytic domain, the cystein-rich do-
main, and the spacer domain. These mutations have 2 func-
tional and non exclusive consequences. The first is the direct
loss of catalytic activity. In vitro experiments showed that
the deletion of the carboxy-terminal end of ADAMTS13
Fig. (2). (A) Western-blot analysis of von Willebrand factor (VWF) multimers in a patient with TTP and from normal human
plasma. VWF Multimers were separated by electrophoresis on polyacrylamide gel electrophoresis according to molecular weight and elec-
tro-transferred on a nitrocellulose membrane. Excessive levels of high molecular weight von Willebrand factor (VWF) multimers are ob-
served in the patient with TTP, as a result of severe ADAMTS13 deficiency. Those unusually high molecular weight multimers are not found
in normal human plasma (NHP). (B) Primary structure of ADAMTS13. PS: peptide signal. TSP-1: thrombospondin-1. CUB: complement
(C1r/C1s), uEGF (urchin epidermal growth factor), bone morphogenetic protein (acronym for 3 proteins harbouring these domains).

4 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
gene, including the CUB domains, the TSP-1 motifs and the
spacer domain only mildly reduced the enzyme activity,
whereas the deletion of the desintegrin-like domain results in
a severely decreased altered vWF cleavage. In patients how-
ever, mutations were found all over ADAMTS13 gene, sug-
gesting that all domains are critical for an optimal enzymatic
activity in vivo [24]. The second consequence of these muta-
tions is a defect in the release of ADAMTS13 by their secret-
ing cells, which may occur at the lateral pole of the cells
[13].
So far, no clear correlation could be established between
ADAMTS13 mutations and clinical presentation, suggesting
the involvement of additional factors in TTP pathophysiol-
ogy. These may include infectious processes (extrinsic fac-
tors), but also yet unknown “modifying genes” (intrinsic
factors) which may act in concert with ADAMTS13 in vari-
ous processes such as platelets aggregation, endothelial acti-
vation, or vascular homeostasis. This view was strongly sup-
ported by the analysis of ADAMTS13-deficient mice, in
which intravenous injection of a microbe-derived toxin de-
rived from bacterial pathogens associated with HUS resulted
in a striking syndrome closely resembling human TTP. Im-
portantly, this syndrome only occurred in Casa/Rk strain
mice, which harbor 5 to 10 times higher circulating VWF
multimers. Moreover, the absence of correlation between
circulating VWF multimers concentration and the severity of
TTP led the authors to conclude that additional genes may be
involved in this process [25]. Interestingly, this relevant
model of congenital TTP also allowed demonstrating the
crucial role of VWF in TTP since CASA/Rk/Adamts13-/-
mice in which VWF gene was also invalidated did not de-
velop TTP after shigatoxin administration [26].
ADAMTS13 Deficiency in Acquired TTP
In acquired TTP, ADAMTS13 deficiency usually results
from autoantibodies altering the protein activity, leading to
consider acquired TTP as an autoimmune disease. The effi-
ciency of immunomodulating agents such as rituximab in the
prevention of relapses in patients with persistent anti-
ADAMTS13 antibodies further comforted this view. In 38 to
95 p. cent of cases, anti-ADAMTS13 antibodies display an
inhibitory activity against the enzyme in vitro, as evidenced
by mixing the plasma of a patient with acquired TTP with
normal human plasma, thereby neutralizing the enzyme ac-
tivity of this latter [23] [27]. The wide range of these results
probably reflects the lack of standardization and reproduci-
bility of the assays for ADAMTS13 functional assays. In
patients with no detectable inhibitory anti-ADAMTS13 anti-
bodies, ADAMTS13 deficiency may be related to inhibitory
antibodies which concentration is below the detection
threshold of the current assays, or either to non-neutralizing
Fig. (3). Pathophysiological mechanisms leading to microthrombi formation in TTP. Endothelial cells may be activated by triggering
factors, mostly infections. Bacterial lipopolysaccharide, various cytokines and Fas-ligand may act in concert to activate endothelial cells.
Damaged endothelial cells release high molecular weight VWF multimers, as well as PAF, and express adhesion molecules at their surface.
In patients with a severe ADAMTS13 deficiency, high molecular weight VWF accumulate, which results in platelet hyperadhesiveness and
platelet clumping within microvasculature of various organs including brain, kidneys, digestive tract and heart. LPS: lipopolysaccharide;
TNF: tumor necrosis factor; IL: interleukin; PAF: platelet activating factor; VWF: von Willebrand factor.

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 5
antibodies, which are identified by ELISA [28]. In most
cases, anti-ADAMTS13 antibodies (either inhibitory or non-
inhibitory) are of IgG type. In rare cases, IgA and/or IgM
isotypes were associated with antibodies of IgG isotype, and
may worsen the prognosis [28] [29]. Anti-ADAMTS13 anti-
bodies may decrease ADAMTS13 activity by directly inhib-
iting the catalytic activity, or by decreasing protein concen-
trations through an opsonization process [29]. The inhibition
of ADAMTS13 binding to its endothelium receptor CD36 is
a tempting possible mechanism that still remains to be dem-
onstrated. Anti-ADAMTS13 may recognize various epitopes
among the protein. Antibodies directed against the cystein-
rich and the spacer domains are the most frequently ob-
served. These may be associated with antibodies directed
against CUB domains, the first thrombospondin-1 (TSP-1)
motif, or the region including the catalytic domain, the desin-
tegrin-like domain, and the first TSP-1 domain. More rarely,
these antibodies recognize the TSP-1 motifs 2 to 8, or the
propeptide domain [30]. Anti-ADAMTS13 IgG, as detected
by ELISA, were found in a majority of patients with severe
ADAMTS13 deficiency (35/36 patients, 97 p. 100), but also
in 3 patients with TTP showing a detectable ADAMTS13
activity (11 to 16 p. 100 of normal activity), which suggests
that ELISA sensitivity may be higher than this of functional
ADAMTS13 assays [31]. On the opposite, these antibodies
were not found in 4 patients with a diagnosis of HUS [31].
Those promising results, suggesting a specificity of anti-
ADAMTS13 for acquired TTP, need however confirmation
on larger series of patients.
Clinical Presentation
TTP is a specific subset of TMA usually characterized by
a microangiopathic hemolytic anemia with a profound pe-
ripheral thrombocytopenia, fever, central nervous system
manifestations, and renal failure [5]. However, these features
are complete only in 40 p. cent of cases; in other instances,
TTP may be revealed by only a bicytopenia without organ
involvement. Indeed, the only association of a microan-
giopathic hemolytic anemia with a peripheral thrombocy-
topenia should alert clinicians for this diagnosis. Importantly,
reports clearly emphasized that TTP may be underestimated
and under-recognized, particularly in children. As a general
rule, TTP should be systematically suspected in children
with a diagnosis of peripheral cytopenia, particularly if asso-
ciated with any organ involvement. In patients with an ap-
parent diagnosis of idiopathic thrombocytopenic purpura or
Evan’s syndrome not responding to usual therapies, the iden-
tification of schistocytes on repeated blood smears, in asso-
ciation with a severe ADAMTS13 deficiency may allow
revisiting the diagnosis [32].
Central nervous system involvement occurs in 84 to 92 p.
cent of cases and was shown to be associated with a worse
prognosis by some groups [33]. It may result in confusion,
obnubilation, headache, somnolence, coma and seizure. A
focal sensitive or motor deficiency, dysarthria, or aphasia
may also be observed. Brain magnetic resonance imagery
may disclose punctiform images in white matter resulting
from ischemia. Thrombosis or hemorrhage may also be ob-
served. Fever is observed in 59 to 98 p. cent of cases. Renal
failure is usually mild and is observed in half of cases. A
mild proteinuria and/or hematuria may also be observed.
Heart involvement may be characterized by thoracic pain,
repolarization or conduction changes, and elevated troponin
Ic, which may reflect left ventricular dysfunction [34]. Di-
gestive involvement, associating vomiting and abdominal
pain with sometimes pancreatitis, or more rarely, lung and
ocular injury, may occur.
Histopathologically, TTP is characterized by the presence
of widespread hyaline thrombi in the terminal arterioles and
capillaries, accounting for organ failure. The high levels of
shear stress in the arterioles and capillaries are believed to be
a critical factor in determining the distribution of thrombosis
in TTP. Autopsic studies revealed that thrombi involved
most organs, including the brain (mainly cerebral cortex),
heart, spleen, pancreas, adrenal gland and kidney. Thrombi
are composed primarily of platelets and VWF, which con-
trasts with the thrombi of disseminated intravascular coagu-
lopathy or the HUS, which are rather characterized by
prominent fibrin deposits [35].
Acquired TTP
Acquired TTP occurs more frequently in females (3 fe-
males for 2 males), within the fourth decade. The onset of
disease is typically sudden. However, prodromic manifesta-
tions including fatigue, arthralgias, myalgias, abdominal or
lumbar pain, suggesting a flu-like episode are frequently
observed. The incidence of TTP was reported to be of 4
cases per million hab. per year. Acquired TTP could be asso-
ciated with various autoimmune diseases, especially SLE (4
p. 100 of SLE cases) [36]. More generally, acquired TTP is
associated in one third of cases with various manifestations
suggesting an underlying autoimmune process. Antinuclear
antibodies or antibodies directed against CD36 are observed
in up to two third of cases [37] [38]. Finally, questioning the
patients with an acquired TTP allow sometimes evidencing a
history of autoimmune disease in other family members.
These observations, arising from series of adult patients or
either from more limited series of pediatric cases [39], highly
suggest that patients with acquired TTP have a trend to de-
velop autoimmune manifestations. In addition, multiple stud-
ies focused on the higher prevalence of acquired TTP in par-
ticular ethnical groups, such as Afro-Caribbeans [27] [38]
[40], suggesting the existence of susceptibility genes in-
volved in the loss of tolerance of the immune system towards
ADAMTS13. The report of acquired TTP in two twin sisters
at 23 and 24-year old further supports this hypothesis [41].
Whether anti-ADAMTS13 antibodies have a prognostic
value in TTP is the matter of intensive research. The identifi-
cation of inhibitory anti-ADAMTS13 antibodies was associ-
ated with a longer time to platelet count recovery and larger
volumes of plasma, in relation with more frequent early re-
lapses during intensive treatment or during the decrease of
plasma exchange sessions [42] [43] [44]. In addition, a high
titre of anti-ADAMTS13 antibodies (either inhibitory or non-

6 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
inhibitory) as evidenced by ELISA was correlated with a
persistently decreased ADAMTS13 activity. Interestingly,
this latter condition could be associated with a relapse risk of
50 p. cent, whereas patients who normalize ADAMTS13
activity may relapse in less than 10 p. cent of cases [29] [45].
Indeed, the study of ADAMTS13 activity after complete
remission achievement may have an important prognostic
value in evaluating the probability of relapse [46] [45]. Mul-
ticentre, large prospective studies with standardized assays
should further clarify the prognostic value of neutralizing
and non-neutralizing anti-ADAMTS13 antibodies and draw-
ing definitive conclusions.
Congenital TTP
Congenital TTP, previously termed Upshaw-Schulman
syndrome, is an autosomal recessive disorder usually ob-
served in children and newborns. Consequently, probands
may be either male or female and parents (who may be con-
sanguineous) are healthy with a mild decrease in ADAMTS13
activity. Congenital TTP may be revealed by cytopenias or
jaundice in the neonatal period (about 75 p. cent of cases) or
later, usually in the first decade [24]. Some congenital TTP
may be revealed in adulthood [47], particularly within a con-
text of pregnancy. Clinical manifestations typically include
anemia, thrombocytopenia, and organ involvement of vari-
able severity. In neonates, unexplained hemolysis and throm-
bocytopenia sometimes result in exsanguino-trans-fusion
sessions. Patients frequently experience repeated relapses at
varying intervals that are frequently triggered by non specific
infectious processes, including flu-like episodes. Initially,
TTP episodes may resolve completely; however, when mul-
tiple relapses occur, those patients with a chronic relapsing
form may develop a chronic renal failure or ischemic lesions
in brain. In other instances, patients may present with mod-
erate chronic hemolysis and thrombocytopenia. A history of
hemolysis or thrombocytopenia may be found in other fam-
ily members.
Treatment and Outcome
Plasmatherapy
Without treatment, acquired TTP is almost always fatal,
whereas under rigorous management, up to 80 p. cent of pa-
tients recover. Treatment is based on therapeutical plasma
exchanges [48] [49], which have to be started as soon as the
diagnosis is established or even suspected. If plasma ex-
changes cannot be performed immediately, high dose plasma
infusions should be started in emergency until plasma ex-
change availability [50]. Plasma exchanges allow bringing
large volumes of plasma that will supply ADAMTS13 defi-
ciency. Plasma exchanges may have additional therapeutical
effects such as removal of anti-ADAMTS13 antibodies or of
high molecular weight VWF from plasma; however, the im-
portance of these latter in patients’ improvement remains
elusive. Plasma exchanges are performed daily until durable
platelet recovery. The rapid or progressive decrease in
plasma exchanges sessions remains a matter of debate. In 30
to 40 p. cent of cases, patients experience one or multiple
relapses after the initial episode, which require to resume
intensive plasmatherapy. The period between relapses may
range from days to many years. In 10 p. cent of cases, TTP is
unresponsive to plasma exchanges and requires more inten-
sive plasmatherapy and/or immunomodulatory therapies.
Patients with congenital TTP are efficiently treated with
plasma infusions. In patients with chronic relapsing forms or
persistent hemolysis and thrombocytopenia, prophylactic
infusions every 2 to 3 weeks may efficiently control the dis-
ease and prevent serious complications. Other patients may
maintain normal or mildly subnormal platelet counts and
require plasma infusion only during intermittent acute exac-
erbations.
Immunomodulatory Therapies
The view that acquired TTP is an autoimmune dis-
ease incites the use of immunomodulatory therapies. Steroids
are frequently proposed at the acute phase of the disease, in
association with plasma exchanges, but their efficacy was
not evaluated on controlled studies. Other immunomodula-
tory agents such as cyclosporin A, azathioprine, or vincris-
tine were also used efficiently in single cases or small series
of patients. Patients with a refractory, threatening disease
may be efficiently treated with pulses of cyclophosphamide
[51].
These last years, the chimeric monoclonal antibody ri-
tuximab, directed against CD20 antigen on B lymphocytes,
became the most frequently used immunomodulatory agent.
Indeed, patients with a refractory disease or showing inter-
mittent relapses were reported to be be efficiently treated
with high response rates, along with an increase in
ADAMTS13 activity and a decrease in anti-ADAMTS13
antibodies [52] [53]. However, the use of rituximab may not
systematically hamper long term relapses. Ongoing large,
prospective studies are attempting to specify the schedule of
rituximab administration in an order to reduce the number of
plasma exchanges, which are associated with significant
side-effects.
HEMOLYTIC UREMIC SYNDROME
Two forms of HUS are classically identified. The first
occurs after an intestinal infection resulting from a shiga-
toxin-producing Escherichia coli (STEC) strain; it is there-
fore termed STEC-associated HUS or diarrhea-associated
HUS (D+ HUS), and represents the most common form of
HUS (for an extensive review, see [54]). The second form,
termed atypical HUS, occurs out of a context of STEC infec-
tion and accounts for 5 to 10 p. cent of all cases of the disor-
der. It was associated with congenital or, more rarely, ac-
quired complement pathway dysfunctions [55]. TMA with
features of atypical HUS were also reported in patients with
abnormalities in cobalamins metabolism, or either in associa-
tion with various conditions such as HIV infection, autoim-
mune diseases, cancers and pregnancy. These latter are
thought to act as triggers causing endothelial cell activation
and injury, probably within a context of genetic susceptibil-
ity [55].
Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 7
Pathophysiology
STEC-Associated HUS
Toxins released by shigatoxin-producing microorgan-
isms, most commonly Escherichia coli O157:H7 (shigatox-
ins 1 and 2) and Shigella dysenteriae serotype 1 (shigatoxin)
strains, are directly involved in D+ HUS pathophysiology
since their injection to non human primate leads to manifes-
tations mimicking HUS in a dose-dependent manner [56].
Shigatoxin is composed of one A subunit of 33 kDa and 5 B
subunits of 7.7 kDa each. Most E. coli O157:H7 carry the
gene encoding shigatoxin 2, and about two-thirds have the
gene encoding shigatoxin 1. Other strains have also been
involved: E. coli O111: H-, O26: H11 and O103: H2. Bacte-
rial agents ingested from contaminating sources proliferate in
the intestinal lumen and adhere to mucosal epithelial cells of
the colon. Shigatoxins damage the underlying tissue and
vasculature and cause bloody diarrhea. The lesions are po-
tentiated by neutrophils, which are recruited into the dam-
aged colon and activated by IL-8 and other chemokines. Shi-
gatoxins cross the intestinal barrier and are transported by
neutrophils, monocytes and platelets in blood flow onto renal
microcirculation. The toxins stimulate the release of TNF- ,
IL-1 and IL-6 from monocytes and renal glomerular and
tubular epithelial cells, which up-regulate the expression of
shigatoxins receptors called globotriaosylceramide and gal-
abiosylceramide.
Shigatoxins bind to their receptors through B subunits on
glomerular capillary endothelial cells, mesangial cells, and
glomerular and tubular epithelial cells. After internalization,
subunit A of shigatoxins inhibits 28S ribosomal subunit,
thereby inhibiting protein synthesis machinery. This process
leads to endothelial cell apoptosis and endothelial injury.
This damage is potentiated by the monocytes and neutrophils
that infiltrate the glomeruli in response to the secreted
chemokines such as IL-8 and fractalkine and the production
of monocyte chemoattractant protein 1 by renal cells. Renal
endothelial cell injury results in the expression of high mo-
lecular weight VWF multimers and adhesion molecules,
such as P-selectin, PECAM-1 (platelet-endothelial-cell adhe-
sion molecule 1) and vitronectin ( V 3 integrin) receptors.
Damaged endothelial cells also express high levels of tissue
factor, plasma activator inhibitor type 1 (PAI-1) and D-
dimers. All these features lead to a local prothrombotic state
resulting in increased platelet adhesiveness with fibrin-rich
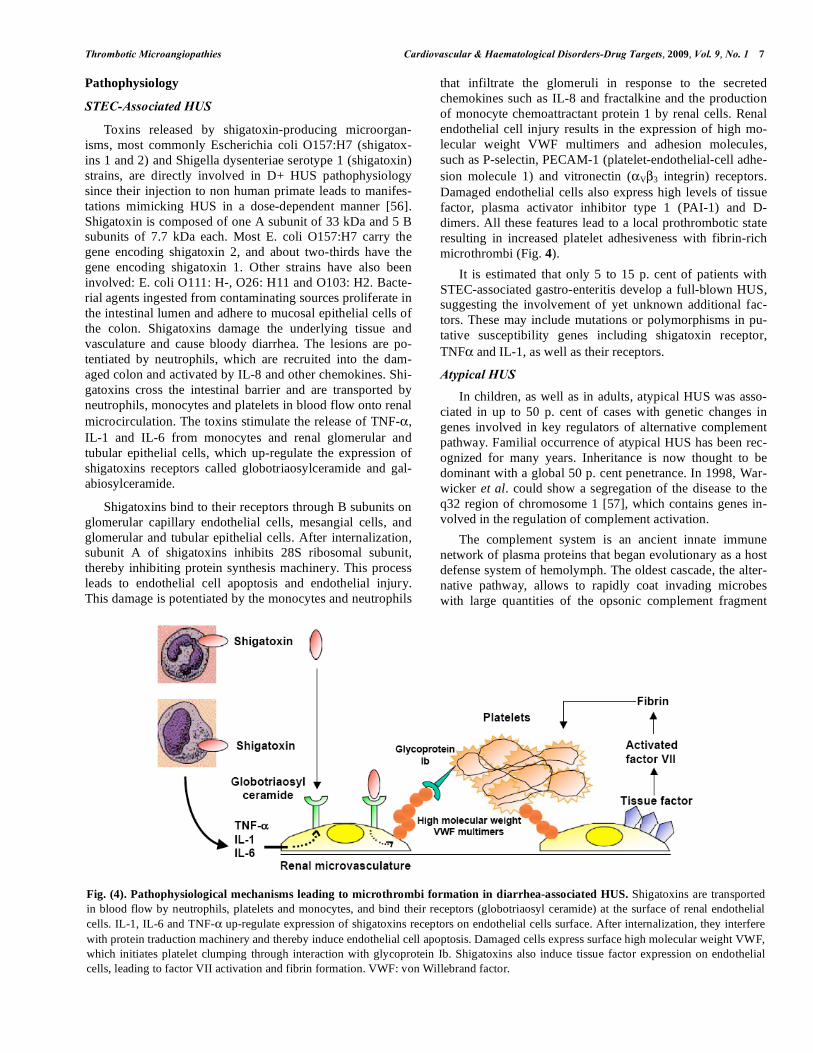
microthrombi (Fig. 4).
It is estimated that only 5 to 15 p. cent of patients with
STEC-associated gastro-enteritis develop a full-blown HUS,
suggesting the involvement of yet unknown additional fac-
tors. These may include mutations or polymorphisms in pu-
tative susceptibility genes including shigatoxin receptor,
TNF and IL-1, as well as their receptors.
Atypical HUS
In children, as well as in adults, atypical HUS was asso-
ciated in up to 50 p. cent of cases with genetic changes in
genes involved in key regulators of alternative complement
pathway. Familial occurrence of atypical HUS has been rec-
ognized for many years. Inheritance is now thought to be
dominant with a global 50 p. cent penetrance. In 1998, War-
wicker et al. could show a segregation of the disease to the
q32 region of chromosome 1 [57], which contains genes in-
volved in the regulation of complement activation.
The complement system is an ancient innate immune
network of plasma proteins that began evolutionary as a host
defense system of hemolymph. The oldest cascade, the alter-
native pathway, allows to rapidly coat invading microbes
with large quantities of the opsonic complement fragment
Fig. (4). Pathophysiological mechanisms leading to microthrombi formation in diarrhea-associated HUS. Shigatoxins are transported
in blood flow by neutrophils, platelets and monocytes, and bind their receptors (globotriaosyl ceramide) at the surface of renal endothelial
cells. IL-1, IL-6 and TNF- up-regulate expression of shigatoxins receptors on endothelial cells surface. After internalization, they interfere
with protein traduction machinery and thereby induce endothelial cell apoptosis. Damaged cells express surface high molecular weight VWF,
which initiates platelet clumping through interaction with glycoprotein Ib. Shigatoxins also induce tissue factor expression on endothelial
cells, leading to factor VII activation and fibrin formation. VWF: von Willebrand factor.

8 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
C3b (Fig. 5). This process is facilitated by an amplification
loop that results in the deposition of several million mole-
cules of C3b on bacteria within a few seconds. Complement
activation may also occur on altered self-tissues, such as on
cells undergoing apoptosis and at sites of injury and infec-
tion. To prevent excessive production and deposits of C3b,
the alternative pathway is finely regulated by proteins that
prevent C3 activation. Indeed, heterozygous mutations (hap-
loinsufficiency) in these regulators predisposes humans to
HUS [58].
Complement Proteins Implicated in Atypical HUS
The first complement protein associated with atypical
HUS was factor H, a 150-kDa plasma protein composed of
20 short consensus repeats of 60 amino-acids each. Factor H
normally protects host cells from accidental damage by the
alternative complement pathway by displacing Bb from C3b,
thereby exposing C3b to cleavage and inactivation by factor
I. Mutations in the factor H gene in atypical HUS have now
been widely described. More than 100 disease-associated
mutations are reported in the factor H-HUS mutations data-
base (http://www.FH-HUS.org). Twenty to 30 p. cent of pa-
tients with atypical HUS were reported to harbour heterozy-
gous mutations in factor H [59]. Missense mutations, dele-
tions and frame shifts in the factor H gene have been identi-
fied in patients and their relatives. Most mutations cluster in
short consensus repeat number 20 of factor H gene in the C-
terminal end of the protein, which disrupts a heparine-
binding site involved in factor H binding to host surfaces. In
most cases, they consist in missense mutations and are asso-
ciated with normal levels of circulating factor H. In the re-
maining cases, they result in either a truncated protein or
impaired secretion of the protein and thus cause a 50 p. cent
reduction in plasma levels of factor H. A mouse model of
HUS designed to mirror human mutations in factor H con-
firmed that the binding of factor H in anionic targets such as
heparine on endothelial cells surface had a crucial impor-
tance since suppression of this function resulted in the occur-
rence of HUS features [60]. Of note, mutations were also
observed in genes harbouring sequence homologies with
factor H gene (CFHR [complement factor H-related]1 and
CFHR3), as well as fusion genes between factor H and
CFHR1. An acquired dysfunction in factor H related to anti-
factor H antibodies of IgG type was also reported in 3 pediat-
ric cases of atypical HUS [61]. Those antibodies are usually
associated with mutations of CFHR1.
Further proteins involved in complement regulation, such
as factor I or CD46/MCP (membrane cofactor protein) were
also found mutated in atypical HUS [62] [63] [64] [65]. Mu-
tations in factor I were reported in < 4 p. cent to 13 p. cent of
cases. In 30 p. cent of cases, this latter is associated with
additional mutations of complement components or with
anti-factor H antibodies. Mutations in CD46/MCP were re-
ported in 13 p. cent of cases [59]. Mutations in the gene en-
coding factor B were found to enhance formation of the al-
ternative C3 convertase C3bBb or increase resistance to in-
activation [66]. More recently, heterozygous mutations in
complement C3 were identified in 11 patients and 3 relatives
with atypical HUS. These were heterozygous missense or
nonsense mutations, which resulted in most cases in a gain of
function of complement activation through a reduced interac-
tion with MCP or factor H (the major inhibitors of the alter-
native complement pathway) [67].
The specific role of these abnormalities in the occurrence
of HUS features still remains hypothetical. They could result
Fig. (5). Key regulators of alternative complement pathway involved in atypical HUS. Mutations in factor H, factor I, CD46/MCP
(membrane cofactor protein), as well as auto-activating mutations in complement factor B and C3 were reported. Factor H dysfunction may
also involve autoantibodies. It is hypothesized that those mutations result in excessive complement activation, with renal endothelial cells
damage and hyperexpression of tissue factor, fibrin formation and thrombi formation. : mutated protein.

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 9
in an excessive activation of complement pathway mediated
by infectious agents or immune complexes, with a subse-
quent release of excessive amounts of complement compo-
nents such as C5 and membrane attack complex. As a conse-
quence, damaged renal endothelial cells may express pro-
coagulant proteins including local tissue factor with factor
VIII binding and activation, leading to the generation of
thrombin and fibrin polymers (Fig. 5). Noteworthy, results
obtained from patients and analysis of animal models clearly
emphasize that atypical HUS appears more and more as a
multigenic disease involving multiple complement genes in
an individual patient. Therefore, those mutations must be
considered to be predisposing rather than causative, as a
trigger factor (such as infections or a pregnancy) is necessary
to initiate the disease [58].
Interestingly, factor H dysfunctions were also reported in
other diseases in which this protein may have a role in pro-
tecting cells against various insults. Indeed, age-related
macular degeneration was associated with a polymorphism
in short consensus repeat 7 of factor H [68], whereas mem-
branoproliferative glomerulopathies were associated with a
complete deficiency in factor H. Mutations in factor H, but
also in factor I and CD46/MCP were reported in HELLP
syndrome [69], suggesting that this disorder may be an addi-
tional form of TMA with complement dysfunction as risk
factors.
Streptococcus Pneumoniae-Associated HUS
HUS associated with a pneumococcal infection is a spe-
cific form of HUS, resulting from the expression of Thom-
sen-Friedenreich antigen at the surface of erythrocytes, endo-
thelial cells, and glomerules. This antigen, which is normally
covered by sialic acid, is exposed by the pneumococcal-
secreted neuraminidase, and subsequently recognized by
circulating IgM, leading to platelet aggregation with endo-
thelial and glomerular lesions. Consequently, plasmatherapy
is usually contra-indicated in this form of HUS since IgM
provided by plasma infusions may exacerbate the disease.
HUS Associated with Cobalamin Disorders
Atypical HUS was associated with dysfunctions of
cobalamin metabolism disorders. Cobalamin (vitamin B12)
derivatives, methylcobalamin and adenosylcobalamin, par-
ticipate as cofactors for the enzymes 5-methyltetrahydro-
folate-homocysteine methyltransferase and methylmalonyl-
CoA mutase. These enzymes are involved in the remethyla-
tion of homocysteine to methionine and in the conversion of
L-methylmalonyl-CoA to succinate, respectively. Comple-
mentation studies subgroup Cbl disorders from cblA to cblH.
CblC complementation group is the principal inborn error of
Cbl metabolism associated with atypical HUS. Renal com-
plications of CblC disease are thought to be mainly due to
hyperhomocysteinemia-induced damage to glomerular endo-
thelium [70].
Clinical Presentation
D+ HUS represents more than 90 p. cent of HUS cases in
children before 3 year-old; therefore, any child presenting for
the first time with a TMA syndrome, but who does not have
diarrhea, could still be infected with an STEC. Those pa-
tients should thus be investigated for the presence of an
STEC by microbiological analysis of the stools and urines.
The incidence of diagnosed E. coli O157:H7 infections is
greater among rural than urban populations, probably be-
cause of greater exposure to animals excreta. Transmission
from cattle to people might be airborne. D+ HUS may occur
within an epidemic context, mostly in the summer and
autumn. In Buenos-Aires (Argentina) where STEC infections
are endemic, HUS has a very high incidence. Bacterial
strains can contaminate unpasteurized milk or dairy prod-
ucts, ground beef, insufficiently cooked food, as well as mu-
nicipal or swimming water. Diarrhea occurs 2 to 12 days
later, and becomes bloody after 1 to 3 days. Abdominal pain
may be intense and greater than is generally observed in
other forms of bacterial gastroenteritis. Defecation may also
be painful.
Cerebral manifestations are important determinants of
morbidity and mortality. They can be observed in 50 p. cent
of cases. Analysis of blood pressure and metabolic parame-
ters on admission did not predict which child would exhibit
cerebral signs. During the course of the illness however,
children with cerebral involvement had more severe azote-
mia, lower minimum sodium concentrations and required
more dialysis [71]. Cerebral magnetic resonance imaging
(MRI) and computerized tomography may evidence features
of cerebral oedema within parieto-occipital white matter,
suggestive of posterior leuko-encephalopathy. These features
were more frequently observed in patients with severe high
blood pressure. Patients with seizure and/or coma may dis-
play ischemic lesions of basal ganglia with hemorrhagic in-
farction [72].
Other non-renal complications such as cardiac dysfunc-
tion, intestinal complications including perforation and ne-
crosis, and pancreatitis have been reported more rarely.
Treatment and Outcome
HUS management systematically requires a symptomatic
treatment, including haemodialysis, control of renin-depen-
dent high blood pressure with angiotensin convertase inhibi-
tors or angiotensin receptors antagonists. In D+ HUS, intra-
venous rehydratation with isotonic crystalloid and mainte-
nance fluid provide optimum nephroprotection. Plasmather-
apy does not seem to modify the prognosis, which remains
good under symptomatic treatment. Antibiotics, antimotility
agents or narcotics were associated with a worsening of the
disease, and should thus be avoided. Non-steroidal anti-
inflammatory agents, by diminishing renal blood flow,
should not be used either. Prognosis of D+ HUS is usually
good, and end stage renal failure or death are observed in up
to 12 p. cent of cases, with 25 p. cent of survivors demon-
strating long term renal sequelae [73].
Atypical HUS is typically characterized by a high mortal-
ity rate (54 p. cent). About half of survivors experience re-
lapses, and over one third require long-term dialysis. From
pediatric series, children with factor H or factor I mutations

10 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
develop relapses leading rapidly to end-stage renal failure
and/or death. Factor H mutations were associated with the
most severe prognosis, since 60 p. cent of patients reached
end stage renal failure or died within <1 year. Half of pa-
tients with factor I mutation have a rapid evolution to end
stage renal failure, and half recover. Patients with factor H or
factor I mutations usually relapse after renal transplantation.
Patients with CD46/MCP mutations have a better prognosis
characterized by a relapsing course without end stage renal
failure. Moreover, the rate of relapse after kidney transplan-
tation is low in this group [74].
Plasma exchanges were reported to have a beneficial
effect in one third of children from all groups except for pa-
tients harbouring MCP mutations [74]. In atypical HUS with
factor H mutations, kidney and liver transplantations were
intended, but this procedure is associated with a significant
mortality risk [75]. Targeted therapies aimed at supplying the
abnormal proteins or inhibiting complement pathway should
be evaluated in the forthcoming years.
HUS associated with cobalamin disorders require par-
enteral administration of hydroxocobalamin [70].
OTHER TMA SYNDROMES
Pregnancy and Post-Partum
TMA occurring in the setting of pregnancy and post-
partum may display features of acquired or congenital TTP,
as well as HUS or HELLP syndrome. Indeed, pregnancy
may reveal ADAMTS13 or complement dysfunctions result-
ing from gene mutations [69]. HELLP syndrome needs to be
identified since it implies a foetal extraction, whereas TTP
may be efficiently treated with only plasma exchanges. Liver
involvement disseminated intravascular coagulation and de-
tectable ADAMTS13 activity, usually higher than 20 p. cent,
which are typical features in HELLP syndrome but not in
TTP, may help to distinguish both diseases. Severe eclamp-
sia and HELLP syndrome may result from a maternal endo-
thelium dysfunction mediated by an excessive release of
both placenta-derived soluble VEGF (vascular-endothelium
growth factor) receptor and endoglin. This latter is a circulat-
ing transforming growth factor (TGF)- 1 co-receptor which
impairs binding of TGF- 1 to its receptor. In concert with
soluble VEGF receptor, endoglin may inhibit downstream
signalling including effects on activation of eNOS (endothe-
lial nitric oxyde synthetase) and vasodilatation, and thereby
induce placental hypoperfusion.
Hematopoietic Stem Cell Transplantation
Hematopoietic stem cell transplantation (HSCT)-
associated TMA was initially considered as a particular form
of TTP with a worse prognosis due to treatment refractori-
ness. ADAMTS13 activity was found consistently normal in
this form of TMA. Consequently, this latter now tends to be
individualized as a specific form of TMA with specific
pathophysiology and prognosis. HSCT-associated TMA is
favoured by numerous initiating factors, which include total
body irradiation in transplant conditioning, infections, medi-
cations such as calcineurin inhibitors, as well as graft versus
host disease grade 2 to 4 [76]. The response to plasma ex-
change is disappointing, and the management should include
as much as possible the treatment of triggering factors. Vari-
ous studies have reported the efficiency of defibrotide, which
is a single strand polyribonucleotide obtained from mammal
DNA. This latter may protect endothelial cells from TNF- -
associated cell death.
Cancers
Stomach, breast and prostate cancers are the most typical
malignancies providing a TMA syndrome. Cancer-associated
TMA onset is usually insidious. Dyspnea, wasting, severe
disseminated intravascular coagulopathy with dacryocytes
and massive erythromyelemia are specific features [77].
Bone marrow investigations frequently display extrahema-
topoietic, metastatic cells. TMA pathophysiology still re-
mains unclear. Metastatic micro-emboli may be involved in
the occlusion of microvessels, thereby inducing erythrocytes
fragmentation and platelet activation. Cytokines such as
TNF may also participate to endothelial agression. In this
context, ADAMTS13 activity is usually measurable and
higher than 20 p. cent. Rare cases of severe acquired
ADAMTS13 deficiency were observed; these latter may
correspond to a paraneoplastic form of TTP (Oberic et al.,
submitted). Prognosis of cancer-associated TMA is very
poor, and depends on treatment responsiveness of the under-
lying malignancy.
Medications
A large number of drugs were associated with TMA,
including antiplatelet agents, antineoplastic drugs and qui-
nine.
TTP with a documented antibody-mediated severe
ADAMTS13 deficiency was reported in a small fraction of
patients treated with ticlopidine, an inhibitor of one of the
platelet adenosine diphosphate receptors, and clopidogrel,
the structurally similar agent [78] [79]. The estimated inci-
dence of ticlopidin-associated TTP is 1 per 1600 to 5000
patients treated, with a disease onset that ranged from 2 to 12
weeks following treatment initiation [80]. Though no clopi-
dogrel-associated cases were initially observed among
20,000 closely monitored patients treated in phase 3 clinical
trials and cohort studies, patients were found to develop ac-
quired TTP within the first two weeks following drug intake,
suggesting a possible causal relationship. The mechanism
leading to anti-ADAMTS13 antibodies production remains
unknown. It is not excluded however that ticlopidin may
induce the development of anti-ADAMTS13 autoimmune
reaction against ADAMTS13 in a mode analogous to the
development of anti-red cell antibodies in association with
the use of alpha-methyldopa. Additionally, ticlopidin was
reported to disrupt production of extracellular matrix com-
ponents critical for microvascular endothelial cell integrity
with induction of apoptosis [81]. Treatment with plasma
exchange usually allows obtaining resolution of TTP in 77 to
84 p. cent of cases [80]. Relapses may occur in clopidogrel-
associated TTP [79].

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 11
Table 1. To a molecular-based classification of TMA syn-
dromes
Severe ADAMTS13 deficiency (TTP)*:
1. TTP + ADAMTS13 mutations
2. Autoimmune TTP :
+ HIV
+ Antiplatelet agents
+ Cancers
+ Pregnancy
“Idiopathic”
Detectable ADAMTS13 activity (HUS):
1. HUS + enteropathogenic bacteria
2. Atypical HUS + complement dysfunction :
Mutations: factor H, CFHR, FI, MCP/CD46, factor B, C3
Fusion genes between factor H and CFHR1
Auto-Abs: Anti-factor H antibodies
3. Atypical HUS + S. pneumoniae
4. Atypical HUS + Cobalamin metabolism disorders
Detectable ADAMTS13 activity (others):
1. Other TMA syndromes:
+ HIV (often AIDS)
+ Cancer (often disseminated)
+ Connective tissue disease
+ Medications
« Idiopathic »
2. HELLP Syndrome
+ sVEGF-R1, sEndoglin and/or complement dysfunction
3. Malignant AHT, CAPS, type II HIT, VOD, severe DIC
* includes rare cases of TTP with decreased but detectable (10-20%) ADAMTS13
activity and anti-ADAMTS13 antibodies [31]. CFHR : complement factor H-related
genes ; HIV: human immunodeficiency virus ; AIDS: acquired immunodeficiency
syndrome. HELLP: hemolysis, elevated liver enzymes, low platelet count. AHT: arte-
rial hypertension. sVEGF: soluble form of VEGF (vascular-endothelium growth fac-
tor); sEndoglin: soluble endoglin; CAPS: catastrophic antiphospholipid syndrome ;
HIT: heparine-induced thrombocytopenia; VOD: veno-occlusive disease; DIC : dis-
seminated intravascular coagulation.
Multiple antineoplastic drugs were associated with TMA.
Particularly, mitomycin C induces TMA with a frequency
that ranges from less than 2 p. cent of cases to 10 p. cent, and
may be dose-dependent. The specific pathophysiological
mechanisms may involve a drug-mediated endothelial injury.
Mitomycin-associated TMA typically associates high blood
pressure with lung oedema. By contrast, central nervous in-
volvement and fever are rarely observed. Response to plasma
exchanges is usually poor. More recently, inhibitors of
VEGF, aimed at inhibiting tumor vascularization, were asso-
ciated with nephritic-range proteinuria, high blood pressure
and TMA [82], suggesting a major role for VEGF in vascular
homeostasis.
HIV Infection
TMA is more frequently observed in HIV-infected pa-
tients than in non-infected individuals [83]. However, HIV-
associated TMA incidence decreased from 1.4 to 0.3 p. cent
since the era of antiretroviral therapies [84] [85]. When
compared to HIV-positive patients without TMA, patients
with HIV-associated TMA have a lower CD4+ T cell count, a
higher HIV RNA viral load and a higher frequency of oppor-
tunistic infections. TMA may result from distinct patho-
physiological mechanisms. HIV-associated TMA with se-
vere ADAMTS13 deficiency usually have less AIDS-related
complications and their median CD4+ T cell count is higher
than in patients with a detectable ADAMTS13 activity. In
the former, prognosis is comparable to this of non-HIV pa-
tients, whereas in the latter, prognosis is poor despite adapted
treatment [86-89].
CONCLUDING REMARKS
These last years, our knowledge in TMA pathophysiol-
ogy outstandingly improved. Indeed, the evidence of a defi-
ciency in ADAMTS13 in TTP allowed understanding the effi-
ciency of plasmatherapy, which opens perspectives of tar-
geted therapies based on recombinant or plasma-purified
ADAMTS13. In addition, to consider acquired idiopathic
TTP as an autoimmune disease incites to introduce immu-
nomodulatory drugs aimed at reducing the number of plasma
exchange sessions. Atypical HUS may also be treated effi-
ciently with purified proteins or complement pathway modu-
lators. Among these latter, eculizumab is a monoclonal anti-
body directed against C5 component of complement, that was
used efficiently in paroxystical nocturnal hemoglobinuria,
where a deficiency in GPI-anchored proteins lead to an ex-
cessive, complement-mediated, intravascular hemolysis [90].
Altogether, these findings should now allow drawing a
rigorous, pathophysiologically-based, TMA classification
(Table 1), and defining more homogeneous groups of pa-
tients. Obviously, these findings open fascinating perspec-
tives of targeted therapies in the forthcoming years.
THE MEMBERS OF THE REFERENCE CENTRE
FOR THE MANAGEMENT OF THROMBOTIC MI-
CROANGIOPATHIES ARE:
Azoulay Elie = Service de Réanimation Médicale,
Hôpital Saint-Louis, Paris
Bordessoule = Service d’Hématologie, Hôpital
Dominique Dupuytren, Limoges
Buffet Marc = Service d’Hématologie clinique et
de Thérapie Cellulaire, Hôpital
Saint-Antoine, Paris
Bussel Annette = Unité de Clinique Transfusion-
nelle, Hôpital Cochin, Paris
Choukroun Gabriel = Service de Néphrologie, Hôpital
Sud, Amiens
Clabault Karine = Service de Réanimation, CHU de
Rouen

12 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
Coppo Paul = Service d’Hématologie clinique et
de Thérapie Cellulaire, Hôpital
Saint-Antoine, Paris
Daubin Cédric = Service de Réanimation Médicale,
Hôpital de Caen
Devaux Edouard = Service de Néphrologie, Centre
Hospitalier de Pontoise
Frémeaux-Bacchi = Laboratoire d’Immunologie, Hôpi-
Véronique tal Européen Georges Pompidou,
Paris
Galicier Lionel = Service d’Immunopathologie,
Hôpital Saint-Louis, Paris
Gruson Didier = Service de néphrologie, CHU de
Bordeaux
Guidet Bertrand = Service de Réanimation Médicale,
Hôpital Saint-Antoine, Paris
Hamidou Mohamed = Service de Médecine Interne,
Hôpital Hôtel-Dieu, Nantes
Heshmati Farhad = Unité de Clinique Transfusion-
nelle, Hôpital Cochin, Paris
Ifrah Norbert = Service d’Hématologie, CHU Lar-
rey, Angers
Korach Jean-Michel = Service de Réanimation Médicale,
Hôpital Châlons-en-Champagne
Mira Jean-Paul = Service de Réanimation Médicale,
Hôpital Cochin
Monge Matthieu = Service de Néphrologie, Hôpital
Sud, Amiens
Moulin Bruno = Service de Néphrologie, Hôpital
civil, Strasbourg
Mousson Christiane = Service de Néphrologie, CHU de
Dijon
Nivet Hubert = Hôpital Bretonneau, Service de
Néphrologie et Immunologie,
Tours
Ojeda-Uribe Mario = Service de Néphrologie, Hôpital
Emile Muller, Mulhouse
Palcoux = Service de Pédiatrie, CHU de
Jean-Bernard Clermont-Ferrand
Poullin Pascale = Service d’hémaphérèse et
d’autotransfusion, Hôpital la Con-
ception, Marseille
Pourrat Jacques = Service de Néphrologie et Immu-
nologie Clinique, CHU Rangueil,
Toulouse
Pouteilnoble Claire = Service de Néphrologie, Centre
Hospitalier Lyon-Sud, Lyon
Provôt François = Service de Néphrologie, Hôpital
Albert Calmette, Lille
Ramakers Michel = Service de Réanimation Médicale,
Hôpital de Caen
Ribeil Jean-Antoine = Service de Thérapie Cellulaire,
Hôpital Necker-Enfants Malades,
Paris
Ronco Pierre = Service de Néphrologie et de
Dialyses, Hôpital Tenon, Paris
Rondeau Eric = Service de Néphrologie et de
Transplantation, Hôpital Tenon,
Paris
Rossi Jean-François = Service Hématologie et Oncologie
Médicale, Hôpital Lapeyronie,
Montpellier
Saheb Samir = Unité de Thérapie Cellulaire et
d’Hémaphérèse, Hôpital la Pitié-
Salpétrière, Paris
Schlemmer Benoît = Service de Réanimation Médicale,
Hôpital Saint-Louis, Paris
Vernant Jean-Paul = Service d’Hématologie, Hôpital la
Pitié-Salpétrière, Paris
Veyradier Agnès = Service d’Hématologie Bi-
ologique, Hôpital Antoine Béclère,
Clamart
Vigneau Cécile = Service de Néphrologie, Hôpital
Pontchaillou, Rennes
Vincent François = Service de Réanimation Médicale,
Hôpital Avicenne, Bobigny
Winckel Alain = Service de Néphrologie, CHU de
Reims
Wolf Martine = Service d’Hématologie Bi-
ologique, Hôpital Antoine Béclère,
Clamart
Zunic Patricia = Service d’Hématologie, Groupe
Hospitalier Sud-Réunion, la Réun-
ion
REFERENCES
[1] Moschcowitz E. Hyaline thrombosis of the terminal arterioles and
capillaries: a hitherto undercribed disease. Proc. N Y Pathol. Soc.,
1924, 24, 21-4.
[2] Espinosa, G.; Bucciarelli, S.; Cervera, R.; Lozano, M.; Reverter,
J.C.; de la Red, G.; Gil, V.; Ingelmo, M.; Font, J.; Asherson,
R.A.Thrombotic microangiopathic haemolytic anaemia and an-
tiphospholipid antibodies. Ann. Rheum. Dis., 2004, 63(6), 730-6.
[3] Wada, H.; Kaneko, T.; Ohiwa, M.; Tanigawa, M.; Hayashi, T.;
Tamaki, S.N.; Minami, K.; Deguchi, K.; Suzuki, T.; Nakano. In-
creased levels of vascular endothelial cell markers in thrombotic
thrombocytopenic purpura. Am. J. Hematol., 1993, 44(2), 101-5.
[4] Gerritsen, M.E.; Bloor, C.M. Endothelial cell gene expression in
response to injury. Faseb. J., 1993, 7(6), 523-32.
[5] Ridolfi, R.L.; Bell, W.R. Thrombotic thrombocytopenic purpura.
Report of 25 cases and review of the literature. Medicine (Balti-
more), 1981, 60(6), 413-28.
[6] Mitra, D.; Jaffe, E.A.; Weksler, B.; Hajjar, K.A.; Soderland, C.;
Laurence, J. Thrombotic thrombocytopenic purpura and sporadic

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 13
hemolytic-uremic syndrome plasmas induce apoptosis in restricted
lineages of human microvascular endothelial cells. Blood, 1997,
89(4), 1224-34.
[7] Dang, C.T., Magid, M.S.; Weksler, B.; Chadburn, A.; Laurence, J.
Enhanced endothelial cell apoptosis in splenic tissues of patients
with thrombotic thrombocytopenic purpura. Blood, 1999, 93(4),
1264-70.
[8] Moake, J.L.; Turner, N.A.; Stathopoulos, N.A.; Nolasco, L.H.;
Hellums, J.D. Involvement of large plasma von Willebrand factor
(vWF) multimers and unusually large vWF forms derived from en-
dothelial cells in shear stress-induced platelet aggregation. J. Clin.
Invest., 1986, 78(6), 1456-61.
[9] Moake, J.L.; Rudy, C.K.; Troll, J.H.; Weinstein, M.J.; Colannino,
N.M.; Azocar, J.; Seder, R.H.; Hong, S.L.; Deykin, D. Unusually
large plasma factor VIII:von Willebrand factor multimers in
chronic relapsing thrombotic thrombocytopenic purpura. N. Engl.
J. Med., 1982, 307(23):1432-5.
[10] Tsai, H.M. Physiologic cleavage of von Willebrand factor by a
plasma protease is dependent on its conformation and requires cal-
cium ion. Blood, 1996, 87(10), 4235-44.
[11] Furlan, M.; Robles, R.; Lamie, B. Partial purification and charac-
terization of a protease from human plasma cleaving von Wille-
brand factor to fragments produced by in vivo proteolysis. Blood,
1996, 87(10), 4223-34.
[12] Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick,
J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.;
Gruppo, R.; Sarode, R.; Shurin, S.B.; Chandrasekaran, V.; Stabler,
S.P.; Sabio, H.; Bouhassira, E.E.; Upshaw, J.D.; Jr, Ginsburg, D.;
Tsai, H.M. Mutations in a member of the ADAMTS gene family
cause thrombotic thrombocytopenic purpura. Nature, 2001, 413
(6855), 488-94.
[13] Sadler, J.E. Von Willebrand factor, ADAMTS13, and thrombotic
thrombocytopenic purpura. Blood, 2008, 112(1), 11-8.
[14] Xie L, Chesterman CN, Hogg PJ. Control of von Willebrand factor
multimer size by thrombospondin-1. J Exp Med 2001, 193(12):
1341-9.
[15] Chauhan, A.K.; Kisucka, J.; Brill, A.; Walsh, M.T.; Scheiflinger,
F.; Wagner, D.D. ADAMTS13, a new link between thrombosis and
inflammation. J. Exp. Med., 2008, 205(9), 2065-74.
[16] Chauhan, A.K.; Motto, D.G.; Lamb, C.B.; Bergmeier, W.; Dockal,
M.; Plaimauer, B.; Scheiflinger, F.; Ginsburg, D.; Wagner, D.D.
Systemic antithrombotic effects of ADAMTS13. J. Exp. Med.
2006, 203(3), 767-76.
[17] Moore, J.C.; Hayward, C.P.; Warkentin, T.E.; Kelton, J.G. De-
creased von Willebrand factor protease activity associated with
thrombocytopenic disorders. Blood, 2001, 98(6), 1842-6.
[18] Mannucci, P.M.; Vanoli, M.; Forza, I.; Canciani, M.T.; Scorza, R..
Von Willebrand factor cleaving protease (ADAMTS-13) in 123 pa-
tients with connective tissue diseases (systemic lupus erythemato-
sus and systemic sclerosis). Haematologica, 2003, 88(8), 914-8.
[19] Mannucci, P.M.; Canciani, M.T.; Forza, I.; Lussana, F.; Lattuada,
A.; Rossi, E. Changes in health and disease of the metalloprotease
that cleaves von Willebrand factor. Blood, 2001, 98(9), 2730-5.
[20] Furlan, M.; Robles, R.; Galbusera, M.; Remuzzi, G.; Kyrle, P.A.;
Brenner, B.; Krause, M.; Scharrer, I.; Aumann, V.; Mittler, U.; So-
lenthaler, M.; Lämmle, B. von Willebrand factor-cleaving protease
in thrombotic thrombocytopenic purpura and the hemolytic-uremic
syndrome. N. Engl. J. Med., 1998, 339(22), 1578-84.
[21] Tsai, H.M.; Lian, E.C. Antibodies to von Willebrand factor-
cleaving protease in acute thrombotic thrombocytopenic purpura.
N. Engl. J. Med., 1998, 339(22), 1585-94.
[22] Bianchi, V.; Robles, R.; Alberio, L.; Furlan, M.; Lammle, B. Von
Willebrand factor-cleaving protease (ADAMTS13) in thrombocy-
topenic disorders: a severely deficient activity is specific for
thrombotic thrombocytopenic purpura. Blood, 2002, 100(2), 710-3.
[23] Veyradier, A.; Obert, B.; Houllier, A.; Meyer, D.; Girma, J.P. Spe-
cific von Willebrand factor-cleaving protease in thrombotic mi-
croangiopathies: a study of 111 cases. Blood, 2001, 98(6), 1765-72.
[24] Veyradier, A.; Lavergne, J.M.; Ribba, A.S.; Obert, B.; Loirat, C.;
Meyer, D.; Girma, J.P. Ten candidate ADAMTS13 mutations in six
French families with congenital thrombotic thrombocytopenic pur-
pura (Upshaw-Schulman syndrome). J. Thromb. Haemost., 2004,
2(3), 424-9.
[25] Motto, D.G.; Chauhan, A.K.; Zhu, G.; Homeister, J.; Lamb, C.B.;
Desch, K.C.; Zhang, W.; Tsai, H.M.; Wagner, D.D.; Ginsburg, D.
Shigatoxin triggers thrombotic thrombocytopenic purpura in ge-
netically susceptible ADAMTS13-deficient mice. J. Clin. Invest.,
2005, 115(10), 2752-2761.
[26] Chauhan, A.K.; Walsh, M.T.; Zhu, G.; Ginsburg, D.; Wagner,
D.D.; Motto, D.G. The combined roles of ADAMTS13 and VWF
in murine models of TTP, endotoxemia, and thrombosis. Blood,
2008, 111(7), 3452-7.
[27] Vesely, S.K.; George, J.N.; Lammle, B.; Studt, J.D.; Alberio, L.;
El-Harake, M.A.; Raskob, G.E. ADAMTS13 activity in thrombotic
thrombocytopenic purpura-hemolytic uremic syndrome: relation to
presenting features and clinical outcomes in a prospective cohort of
142 patients. Blood, 2003, 102(1), 60-8.
[28] Scheiflinger, F.; Knobl, P.; Trattner, B.; Plaimauer, B.; Mohr, G.;
Dockal, M.; Dorner, F.; Rieger, M. Nonneutralizing IgM and IgG
antibodies to von Willebrand factor-cleaving protease (ADAMTS-
13) in a patient with thrombotic thrombocytopenic purpura. Blood,
2003, 102(9), 3241-3.
[29] Ferrari, S.; Scheiflinger, F.; Rieger, M.; Mudde, G.; Wolf, M.;
Coppo, P.; Girma3, J-P.; Azoulay, E.; Brun-Buisson, C.; Fakhouri,
F.; Mira, J-P.; Oksenhendler, E.; Poullin, P.; Rondeau, E.; Schlei-
nitz, N.; Schlemmer, B.; Teboul, J-L.; Vanhille, P.; Vernant, J-P.;
Meyer, D.; Veyradier, A. Prognostic value of anti-ADAMTS13 an-
tibodies features (Ig isotype, titer and inhibitory effect) in a cohort
of 35 adult French patients undergoing a first episode of thrombotic
microangiopathy with an undetectable ADAMTS13 activity. Blood,
2006.
[30] Klaus, C.; Plaimauer, B.; Studt, J.D.; Dorner, F.; Lammle, B.;
Mannucci, P.M.; Scheiflinger, F. Epitope mapping of ADAMTS13
autoantibodies in acquired thrombotic thrombocytopenic purpura.
Blood, 2004, 103(12), 4514-9.
[31] Rieger, M.; Mannucci, P.M.; Kremer Hovinga, J.A.; Herzog, A.;
Gerstenbauer, G.; Konetschny, C.; Zimmermann, K.; Scharrer, I.;
Peyvandi, F.; Galbusera, M.; Remuzzi, G.; Böhm, M.; Plaimauer,
B.; Lämmle, B.; Scheiflinger, F. ADAMTS13 autoantibodies in pa-
tients with thrombotic microangiopathies and other immunomedia-
ted diseases. Blood, 2005, 106(4), 1262-7.
[32] Schneppenheim, R.; Budde, U.; Oyen, F.; Angerhaus, D.; Aumann,
V.; Drewke, E.; Hassenpflug, W.; Häberle, J.; Kentouche, Kohne,
E.; Kurnik, K.; Mueller-Wiefel, D.; Obser, T.; Santer, R.; Sykora,
K.W. von Willebrand factor cleaving protease and ADAMTS13
mutations in childhood TTP. Blood, 2003, 101(5), 1845-50.
[33] Pene, F.; Vigneau, C.; Auburtin, M.; Moreau, D.; Zahar, J.R.;
Coste, J.; Heshmati, F.; Mira, J.P. Outcome of severe adult throm-
botic microangiopathies in the intensive care unit. Intensive Care
Med., 2005, 31(1), 71-8.
[34] Patschan, D.; Witzke, O.; Duhrsen, U.; Erbel, R.; Philipp, T.; Her-
get-Rosenthal, S. Acute myocardial infarction in thrombotic mi-
croangiopathies--clinical characteristics, risk factors and outcome.
Nephrol. Dial. Transplant., 2006, 21(6),1549-54.
[35] Hosler, G.A.; Cusumano, A.M.; Hutchins, G.M. Thrombotic
thrombocytopenic purpura and hemolytic uremic syndrome are dis-
tinct pathologic entities. A review of 56 autopsy cases. Arch.
Pathol. Lab. Med., 2003, 127(7), 834-9.
[36] Musio, F.; Bohen, E.M.; Yuan, C.M.; Welch, P.G. Review of
thrombotic thrombocytopenic purpura in the setting of systemic lu-
pus erythematosus. Semin. Arthritis Rheum., 1998, 28(1), 1-19.
[37] Tandon, N.N.; Rock, G.; Jamieson, G.A. Anti-CD36 antibodies in
thrombotic thrombocytopenic purpura. Br J Haematol 1994, 88(4),
816-25.
[38] Coppo, P.; Bengoufa, D.; Veyradier, A.; Wolf, M.; Bussel, A.;
Millot, G.A.; Malot, S.; Heshmati, F.; Mira, J.P.; Boulanger, E.;
Galicier, L.; Durey-Dragon, M.A.; Frémeaux-Bacchi, V.; Ramak-
ers, M.; Pruna, A.; Bordessoule, D.; Gouilleux, V.; Scrobohaci,
M.L.; Vernant, J.P.; Moreau, D.; Azoulay, E.; Schlemmer, B.; Gui-
llevin, L.; Lassoued, K. Severe ADAMTS13 deficiency in adult
idiopathic thrombotic microangiopathies defines a subset of pa-
tients characterized by various autoimmune manifestations, lower
platelet count, and mild renal involvement. Medicine (Baltimore),
2004, 83(4), 233-4
[39] Brunner, H.I.; Freedman, M.; Silverman, E.D. Close relationship
between systemic lupus erythematosus and thrombotic thrombocy-

14 Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 Raghupathy and Billett
topenic purpura in childhood. Arthritis. Rheum, 1999, 42(11),
2346-55.
[40] Scully, M.; Yarranton, H.; Liesner, R.; Cavenagh, J.; Hunt, B.;
Benjamin, S.; Bevan, D.; Mackie, I.; Machin, S. Regional UK TTP
Registry: correlation with laboratory ADAMTS 13 analysis and
clinical features. Br. J. Haematol., 2008, 142(5), 819-26 2008.
[41] Studt, J.D.; Hovinga, J.A.; Radonic, R.; Gasparovic, V.; Ivanovic,
D.; Merkler, M.; Wirthmueller, U.; Dahinden, C.; Furlan, M.;
Lämmle, B. Familial acquired thrombotic thrombocytopenic purpu-
ra: ADAMTS13 inhibitory autoantibodies in identical twins. Blood,
2004, 103(11), 4195-7.
[42] Tsai, H.M. High titers of inhibitors of von Willebrand factor-
cleaving metalloproteinase in a fatal case of acute thrombotic
thrombocytopenic purpura. Am. J. Hematol., 2000, 65(3), 251-5.
[43] Zheng, X.L.; Kaufman, R.M.; Goodnough, L.T.; Sadler, J.E. Effect
of plasma exchange on plasma ADAMTS13 metalloprotease activ-
ity, inhibitor level, and clinical outcome in patients with idiopathic
and nonidiopathic thrombotic thrombocytopenic purpura. Blood,
2004, 103(11), 4043-9.
[44] Coppo, P.; Wolf, M.; Veyradier, A.; Bussel, A.; Malot, S.; Millot,
G.A.; Daubin, C.; Bordessoule, D.; Pène, F.; Mira, J.P.; Heshmati,
F.; Maury, E.; Guidet, B.; Boulanger, E.; Galicier, L.; Parquet, N.;
Vernant, J.P.; Rondeau, E.; Azoulay, E.; Schlemmer, B. Prognostic
value of inhibitory anti-ADAMTS13 antibodies in adult-acquired
thrombotic thrombocytopenic purpura. Br. J. Haematol.,
2006,132(1), 66-74.
[45] Peyvandi F, Lavoretano S, Palla R, Feys HB, Vanhoorelbeke K,
Battaglioli T,
[46] Peyvandi, F.; Lavoretano, S.; Palla, R.; Feys, H.B.; Vanhoorelbeke,
K.; Battaglioli, T.; Valsecchi, C.; Canciani, M.T.; Fabris, F.; Zver,
S.; Réti, M.; Mikovic, D.; Karimi, M.; Giuffrida, G.; Laurenti, L.;
Mannucci, P.M. ADAMTS13 and anti-ADAMTS13 antibodies as
markers for recurrence of acquired thrombotic thrombocytopenic
purpura during remission. Haematologica, 2008, 93(2), 232-9.
[47] Mannucci, P.M.; Peyvandi, F. TTP and ADAMTS13, When Is
Testing Appropriate? Hematology Am. Soc. Hematol. Educ. Pro-
gram, 2007, 121-6.
[48] Uchida, T.; Wada, H.; Mizutani, M.; Iwashita, M.; Ishihara, H.;
Shibano, T.; Suzuki, M.; Matsubara, Y.; Soejima, K.; Matsumoto,
M.; Fujimura, Y.; Ikeda, Y.; Murata, M.; Research Project on Ge-
netics of Thrombosis. Identification of novel mutations in
ADAMTS13 in an adult patient with congenital thrombotic throm-
bocytopenic purpura. Blood, 2004, 104(7), 2081-3.
[49] Rock, G.A.; Shumak, K.H.; Buskard, N.A.; Blanchette, V.S.; Kel-
ton, J.G.; Nair, R.C.; Spasoff, R.A. Comparison of plasma exchan-
ge with plasma infusion in the treatment of thrombotic thrombocy-
topenic purpura. Canadian Apheresis Study Group. N. Engl. J.
Med., 1991, 325(6), 393-7.
[50] Bell, W.R.; Braine, H.G.; Ness, P.M.; Kickler, T.S. Improved sur-
vival in thrombotic thrombocytopenic purpura-hemolytic uremic
syndrome. Clinical experience in 108 patients. N. Engl. J. Med.,
1991, 325(6), 398-403.
[51] Coppo, P.; Bussel, A.; Charrier, S.; Adrie, C.; Galicier, L.; Boulan-
ger, E.; Veyradier, A.; Leblanc, T.; Alberti, C.; Azoulay, E.; Le
Gall, J.R.; Schlemmer, B. High-dose plasma infusion versus plas-
ma exchange as early treatment of thrombotic thrombocytopenic
purpura/hemolytic-uremic syndrome. Medicine (Baltimore), 2003,
82(1), 27-38.
[52] Bird, J.M.; Cummins, D.; Machin, S.J. Cyclophosphamide for
chronic relapsing thrombotic thrombocytopenic purpura. Lancet,
1990, 336(8714), 565-6.
[53] Fakhouri, F.; Vernant, J.P.; Veyradier, A.; Wolf, M.; Kaplanski, G.;
Binaut, R.; Rieger, M.; Scheiflinger, F.; Poullin, P.; Deroure, B.;
Delarue, R.; Lesavre, P.; Vanhille, P.; Hermine, O.; Remuzzi, G.;
Grünfeld, J.P. Efficiency of curative and prophylactic treatment
with rituximab in ADAMTS13-deficient thrombotic thrombocyt-
openic purpura: a study of 11 cases. Blood, 2005, 106(6), 1932-7.
[54] Scully, M.; Cohen, H.; Cavenagh, J.; Benjamin, S.; Starke, R.;
Killick, S.; Mackie, I.; Machin, S.J. Remission in acute refractory
and relapsing thrombotic thrombocytopenic purpura following ri-
tuximab is associated with a reduction in IgG antibodies to
ADAMTS-13. Br. J. Haematol., 2007, 136(3), 451-61.
[55] Tarr, P.I.; Gordon, C.A.; Chandler, W.L. Shiga-toxin-producing
Escherichia coli and haemolytic uraemic syndrome. Lancet, 2005,
365(9464), 1073-86.
[56] Zipfel, P.F.; Heinen, S.; Jozsi, M.; Skerka, C. Complement and
diseases: defective alternative pathway control results in kidney
and eye diseases. Mol. Immunol., 2006, 43(1-2), 97-106.
[57] Siegler, R.L.; Pysher, T.J.; Lou, R; Tesh, V.L.; Taylor, F.B.; Jr.
Response to Shiga toxin-1, with and without lipopolysaccharide, in
a primate model of hemolytic uremic syndrome. Am. J. Nephrol.,
2001, 21(5), 420-5.
[58] Warwicker, P.; Goodship, T.H.; Donne, R.L.; Pirson, Y.; Nicholls,
A.; Ward, R.M.; Turnpenny, P.; Goodship, J.A. Genetic studies
into inherited and sporadic hemolytic uremic syndrome. Kidney
Int., 1998, 53(4), 836-44.
[59] Atkinson, J.P.; Goodship, T.H. Complement factor H and the
hemolytic uremic syndrome. J. Exp. Med., 2007, 204(6), 1245-8.
[60] Caprioli, J.; Noris, M.; Brioschi, S.; Pianetti, G.; Castelletti, F.;
Bettinaglio, P.; Mele, C.; Bresin, E.; Cassis, L.; Gamba, S.; Porrati,
F.; Bucchioni, S.; Monteferrante, G.; Fang, C.J.; Liszewski, M.K.;
Kavanagh, D.; Atkinson, J.P.; Remuzzi, G.; International Registry
of Recurrent and Familial HUS/TTP. Genetics of HUS: the impact
of MCP, CFH, and IF mutations on clinical presentation, response
to treatment, and outcome. Blood, 2006, 108(4), 1267-79.
[61] Pickering, M.C.; de Jorge, E.G.; Martinez-Barricarte, R.; Recalde,
S.; Garcia-Layana, A.; Rose, K.L.; Moss, J.; Walport, M.J.; Cook,
H.T.; de Córdoba, S.R.; Botto, M. Spontaneous hemolytic uremic
syndrome triggered by complement factor H lacking surface recog-
nition domains. J. Exp. Med., 2007, 204(6), 1249-56.
[62] Dragon-Durey, M.A.; Frémeaux-Bacchi, V.; Loirat, C.; Blouin, J.;
Niaudet, P.; Deschenes, G.; Coppo, P.; Herman Fridman, W.;
Weiss, L. Heterozygous and homozygous factor h deficiencies as-
sociated with hemolytic uremic syndrome or membranoprolifera-
tive glomerulonephritis: report and genetic analysis of 16 cases. J.
Am. Soc. Nephrol., 2004,15(3), 787-95.
[63] Noris, M.; Brioschi, S.; Caprioli, J.; Todeschini, M.; Bresin, E.;
Porrati, F.; Gamba, S.; Remuzzi, G.; International Registry of Re-
current and Familial HUS/TTP.. Familial haemolytic uraemic syn-
drome and an MCP mutation. Lancet, 2003, 362(9395), 1542-7.
[64] Fremeaux-Bacchi, V.; Dragon-Durey, M.A.; Blouin, J.; Vigneau,
C.; Kuypers, D.; Boudailliez, B.; Loirat, C.; Rondeau, E.; Fridman,
W.H. Complement factor I: a susceptibility gene for atypical
haemolytic uraemic syndrome. J. Med. Genet., 2004, 41(6), e84.
[65] Goodship, T.H.; Liszewski, M.K.; Kemp, E.J.; Richards, A.; Atkin-
son, J.P. Mutations in CD46, a complement regulatory protein, pre-
dispose to atypical HUS. Trends Mol. Med., 2004, 10(5), 226-31.
[66] Kavanagh D, Kemp EJ, Mayland E, Winney RJ, Duffield JS, War-
wick G, et al. Mutations in complement factor I predispose to de-
velopment of atypical hemolytic uremic syndrome. J Am Soc
Nephrol 2005,16(7):2150-5.
[67] Goicoechea de Jorge, E.; Harris, C.L.; Esparza-Gordillo, J.; Car-
reras, L.; Arranz, E.A.; Garrido, C.A.; López-Trascasa, M.;
Sánchez-Corral, P.; Morgan, B.P.; Rodríguez de Córdoba S. Gain-
of-function mutations in complement factor B are associated with
atypical hemolytic uremic syndrome. Proc. Natl. Acad. Sci. USA,
2007, 104(1), 240-5.
[68] Fremeaux-Bacchi V, Miller EC, Liszewski MK, Strain L, Blouin J,
Brown AL, et al. Mutations in complement C3 predispose to de-
velopment of atypical hemolytic uremic syndrome. Blood 2008 in
press.
[69] Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.;
Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Bor-
chardt, J.D.; Gehrs, K.M.; Smith, R.J.; Silvestri, G.; Russell, S.R.;
Klaver, C.C.; Barbazetto, I.; Chang, S.; Yannuzzi, L.A.; Barile,
G.R.; Merriam, J.C.; Smith, R.T.; Olsh, A.K.; Bergeron, J.; Zernant
J.; Merriam, J.E.; Gold, B.; Dean, M.; Allikmets, R. A common
haplotype in the complement regulatory gene factor H (HF1/CFH)
predisposes individuals to age-related macular degeneration. Proc.
Natl. Acad. Sci. USA, 2005, 102(20), 7227-32.
[70] Fakhouri, F.; Jablonski, M.; Lepercq, J.; Blouin, J.; Benachi, A.;
Hourmant, M.; Pirson, Y.; Durrbach, A.; Grunfeld, J.P.; Knebel-
mann, B.; Fremeaux-Bacchi, V. Factor H, membrane cofactor pro-
tein and Factor I mutations in patients with HELLP syndrome.
Blood, 2008.

Thrombotic Microangiopathies Cardiovascular & Haematological Disorders-Drug Targets, 2009, Vol. 9, No. 1 15
[71] Sharma AP, Greenberg CR, Prasad AN, Prasad C. Hemolytic ure-
mic syndrome (HUS) secondary to cobalamin C (cblC) disorder.
Pediatr Nephrol 2007, 22, 2097-103.
[72] Bale JF, Jr., Brasher C, Siegler RL. CNS manifestations of the
hemolytic-uremic syndrome. Relationship to metabolic alterations
and prognosis. Am J Dis Child 1980,134(9):869-72.
[73] Theobald I, Kuwertz-Broking E, Schiborr M, Heindel W. Central
nervous system involvement in hemolytic uremic syndrome
(HUS)--a retrospective analysis of cerebral CT and MRI studies.
Clin Nephrol 2001,56(6):S3-8.
[74] Garg, A.X.; Suri, R.S.; Barrowman, N.; Rehman, F.; Matsell, D.;
Rosas-Arellano, M.P.; Salvadori, M.; Haynes, R.B.; Clark, W.F.
Long-term renal prognosis of diarrhea-associated hemolytic uremic
syndrome: a systematic review, meta-analysis, and meta-
regression. JAMA, 2003, 290(10), 1360-70.
[75] Sellier-Leclerc, A.L.; Fremeaux-Bacchi, V.; Dragon-Durey, M.A.;
Macher, M.A.; Niaudet, P.; Guest, G.; Boudailliez, B.; Bouissou,
F.; Deschenes, G.; Gie, S.; Tsimaratos, M.; Fischbach, M.; Morin,
D.; Nivet, H.; Alberti, C.; Loirat, C.; French Society of Pediatric
Nephrology. Differential impact of complement mutations on clini-
cal characteristics in atypical hemolytic uremic syndrome. J. Am.
Soc. Nephrol., 2007, 18(8), 2392-400.
[76] Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recur-
rence after renal transplantation. Pediatr Transplant 2008.
[77] Roy V, Rizvi MA, Vesely SK, George JN. Thrombotic thrombocy-
topenic purpura-like syndromes following bone marrow transplan-
tation: an analysis of associated conditions and clinical outcomes.
Bone Marrow Transplant 2001,27(6):641-6.
[78] Francis KK, Kalyanam N, Terrell DR, Vesely SK, George JN.
Disseminated malignancy misdiagnosed as thrombotic thrombocy-
topenic purpura: A report of 10 patients and a systematic review of
published cases. Oncologist 2007,12(1):11-9.
[79] Bennett CL, Weinberg PD, Rozenberg-Ben-Dror K, Yarnold PR,
Kwaan HC, Green D. Thrombotic thrombocytopenic purpura asso-
ciated with ticlopidine. A review of 60 cases. Ann Intern Med
1998,128(7):541-4.
[80] Bennett, C.L.; Connors, J.M.; Carwile, J.M.; Moake, J.L.; Bell,
W.R.; Tarantolo, S.R.; McCarthy, L.J.; Sarode, R.; Hatfield, A.J.;
Feldman, M.D.; Davidson, C.J.; Tsai, H.M.. Thrombotic thrombo-
cytopenic purpura associated with clopidogrel. N. Engl. J. Med.,
2000, 342(24):1773-7.
[81] Bennett,C.L.; Kim, B.; Zakarija, A.; Bandarenko, N.; Pandey,
D.K.; Buffie, C.G.; McKoy, J.M.; Tevar, A.D.; Cursio, J.F.;
Yarnold, P.R.; Kwaan, H.C.; De Masi, D.; Sarode, R.; Raife, T.J.;
Kiss, J.E.; Raisch, D.W.; Davidson, C.; Sadler, J.E.; Ortel, T.L.;
Zheng, X.L.; Kato, S.; Matsumoto, M.; Uemura, M.; Fujimura, Y.;
SERF-TTP Research Group.. Two mechanistic pathways for
thienopyridine-associated thrombotic thrombocytopenic purpura: a
report from the SERF-TTP Research Group and the RADAR Pro-
ject. J. Am. Coll. Cardiol., 2007, 50(12), 1138-43.
[82] Mauro, M.; Zlatopolskiy, A.; Raife, T.J.; Laurence, J. Thienopyri-
dine-linked thrombotic microangiopathy: association with endothe-
lial cell apoptosis and activation of MAP kinase signalling cas-
cades. Br. J. Haematol., 2004, 124(2), 200-10.
[83] Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas,
M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx,
P.H.; Gerber, H.P.; Ferrara, N.; Barisoni, L.; Alpers, C.E.; Quag-
gin, S.E. VEGF inhibition and renal thrombotic microangiopathy.
N. Engl. J. Med., 2008, 358(11), 1129-36.
[84] Bell, W.R.; Chulay, J.D.; Feinberg, J.E. Manifestations resembling
thrombotic microangiopathy in patients with advanced human im-
munodeficiency virus (HIV) disease in a cytomegalovirus prophy-
laxis trial (ACTG 204). Medicine (Baltimore), 1997, 76(5), 369-80.
[85] Gervasoni, C.; Ridolfo, A.L.; Vaccarezza, M.; Parravicini, C.;
Vago, L.; Adorni F, et al. Thrombotic microangiopathy in patients
with acquired immunodeficiency syndrome before and during the
era of introduction of highly active antiretroviral therapy. Clin. In-
fect. Dis., 2002, 35(12), 1534-40.
[86] Becker, S.; Fusco, G.; Fusco, J.; Balu, R.; Gangjee, S.; Brennan,
C.; Feinberg, J. HIV-associated thrombotic microangiopathy in the
era of highly active antiretroviral therapy: an observational study.
Clin. Infect. Dis., 2004, 39 Suppl 5, S267-75.
[87] Gadallah, M.F.; el-Shahawy, M.A.; Campese, V.M.; Todd, J.R.;
King, J.W. Disparate prognosis of thrombotic microangiopathy in
HIV-infected patients with and without AIDS. Am. J. Nephrol.,
1996, 16(5), 446-50.
[88] Ahmed, S.; Siddiqui, R.K.; Siddiqui, A.K.; Zaidi, S.A.; Cervia, J.
HIV associated thrombotic microangiopathy. Postgrad. Med. J.,
2002, 78(923), 520-5.
[89] Novitzky, N.; Thomson, J.; Abrahams, L.; du Toit, C.; McDonald,
A. Thrombotic thrombocytopenic purpura in patients with retrovi-
ral infection is highly responsive to plasma infusion therapy. Br. J.
Haematol., 2005, 128(3),373-9.
[90] Malak, S.; Wolf, M.; Millot, G.A.; Mariotte, E.; Veyradier, A.;
Meynard, J.L.; Korach, J.M.; Malot, S.; Bussel, A.; Azoulay, E.;
Boulanger, E.; Galicier, L.; Devaux, E.; Eschwège, V.; Gallien, S.;
Adrie, C.; Schlemmer, B.; Rondeau, E.; Coppo, P.; Azoulay, E.;
Belhocine, R.; Bengoufa, D.; Bordessoule, D.; Buffet, M.; Bussel,
A.; Choukroun, G.; Clabault, K.; Coppo, P.; Daubin, C.; Devaux,
E.; Frémeaux-Bacchi, V.; Galicier, L.; Guidet, B.; Hamidou, M.;
Heshmati, F.; Korach, J.M.; Mira, J.P.; Monge, M. Poullin, P.;
Ramakers, M.; Ribeil, Rondeau, E.; Saheb, S.; {Samy Modeliar},
S.; Schlemmer, B.; Vernant, J.P.; Veyradier, A.; Vincent, F.;
Winckel, A.; M.W. Human immunodeficiency virus-associated
thrombotic microangiopathies: clinical characteristics and outcome
according to ADAMTS13 activity. Scand. J. Immunol., 2008,
68(3), 337-44.
[91] Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socié, G.;
Muus, P.; Röth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.;
Browne, P.; Risitano, A.M.; Hill, A.; Schrezenmeier, H.; Fu, C.L.;
Maciejewski, J.; Rollins, S.A.; Mojcik, C.F.; Rother, R.P.; Luz-
zatto, L. The complement inhibitor eculizumab in paroxysmal noc-
turnal hemoglobinuria. N. Engl. J. Med., 2006, 355(12), 1233-43.