case report/caso clínico
TRANSCRIPT

79
case report/caso clínico
acta obstet ginecol port 2012;6(2):79-82
aBStract
Apert syndrome is a rare genetic disorder characterized by craniosynostosis, midfacial hypoplasia and severe sym-metrical syndactyly (cutaneous and bony fusion) of the hands and feet. Despite being inherited in an autosomal dominant manner, most cases are sporadic, the result of a de novo mutation. The prevalence of this condition at birth is estimated as 15.5 per million (1:65 000). We report the prenatal diagnosis of Apert syndrome associated with increased nuchal translucency and with no ultrasonographic indings of craniosynostosis. An obstetrical ul-trasound performed at 20 weeks of gestation showed ocular hypertelorism and bilateral syndactyly of the hands.
Examination of the remaining fetal structures revealed no further fetal anomalies, in particular no prema-ture fusion of the cranial sutures. Genetic evaluation revealed a S252W mutation in ibroblast growth factor receptor 2, consistent with Apert syndrome.
Fetal autopsy performed at 24 weeks showed a male fetus with symmetric syndactyly of both hands and feet; the skull showed a mild frontal bossing, ocular hypertelorism, depressed nasal bridge and conirmed the absence of premature fusion of the cranial sutures. Prenatal diagnosis of Apert syndrome in sporadic cases can be dificult because the characteristic changes in cranial and orbital shape related to craniosynostosis may not be present until the third-trimester. Syndactyly and abnormalities of the skull shape should lead to the suspicion of Apert syndrome even in the absence of craniosynostosis. This case also suggests that increased nuchal trans-lucency may be associated with this syndrome.
Keywords: Acrocephalosyndactylia; prenatal diagnosis
prenatal diagnosis of apert syndrome without craniosynostosis - case report
diagnóstico pré-natal do síndrome de apert sem craniossinostose – caso clínico
Horácio Azevedo*, Elsa Pereira**, James Anderson**, Adosinda Rosmaninho***, Carla Barbêdo****
* Interno de Ginecologia e Obstetrícia** Assistente de Ginecologia e Obstetrícia
*** Assistente Graduado de Ginecologia e Obstetrícia**** Médica Fetopatologista
Departamento de Ginecologia e ObstetríciaCentro Hospitalar do Alto Ave e Centro de Patologia do Desenvolvimento
introduction
Described by Wheaton in 1894 and later in 1906 by the French physician Apert, acrocephalosyndactyly type I or
Apert syndrome is a rare genetic disorder characterized by craniosynostosis (i.e. premature fusion of one or more cra-nial sutures), craniofacial anomalies, and severe symmetri-cal syndactyly (cutaneous and bony fusion) of the hands and feet. Despite being inherited in an autosomal dominant man-ner, most cases are sporadic, the result of a de novo muta-tion1. The birth prevalence of this condition is estimated as 15,5 per million (1:65 000), the male to female ratio is 1:12.

80
Prenatal detection of the syndrome became possible with the routine prenatal ultrasound screening of fetal anomalies. Once suspected, the diagnosis can be conirmed by DNA analysis of mutations in the ibroblast growth factor receptor 2 gene (FGFR2)3. Prenatal diagnosis of Apert syndrome is established precociously when there is a suggestive familiar history but rather late in the majority of cases because facial and skull abnormalities due to craniosynostosis are often third-trimester ultrasonographic indings.
caSe report
A 34 -year-old woman in her irst pregnancy, with a 35-year-old partner, was referred to our unit for prenatal routine care. In the irst trimester scan, performed at 12 weeks, the fetal crown–rump length and nuchal translucency were 59 mm and 3.7 mm, respectively and nasal bone was present. The esti-mated risk for Down syndrome, after the prenatal biochemical screening [measurement of free β subunit of chorionic gonad-otrophin (FβhCG) and pregnancy-associated plasma protein A (PAPP-A)], was 1/62. An amniocentesis was performed and
Figure 1.
Figure 2.
Figure 3.
Figure 4.
cytogenetic analysis demonstrated a normal male karyotype. A fetal ultrasound performed at 20 weeks of gestation showed ocular hypertelorism (ig. 1) and syndactyly of the hands (ig. 2, 3). Examination of remaining fetal structures, especially in-tracranial, revealed no other fetal anomalies. Although there were no signs of craniosynostosis the couple were informed that the indings were suggestive of Apert syndrome. Molecu-lar analysis of the FGFR2 gene, performed on DNA extracted from the amniocytes, showed a S252W mutation consistent with Apert syndrome. After being informed of the progno-sis, including the probability of low intellectual function and the likely need for multiple cranial and limb surgeries, they
azevedo H, pereira e, anderson J, rosmaninho a, Barbêdo c
81
elected to terminate the pregnancy. This was performed at 24 weeks of gestation.
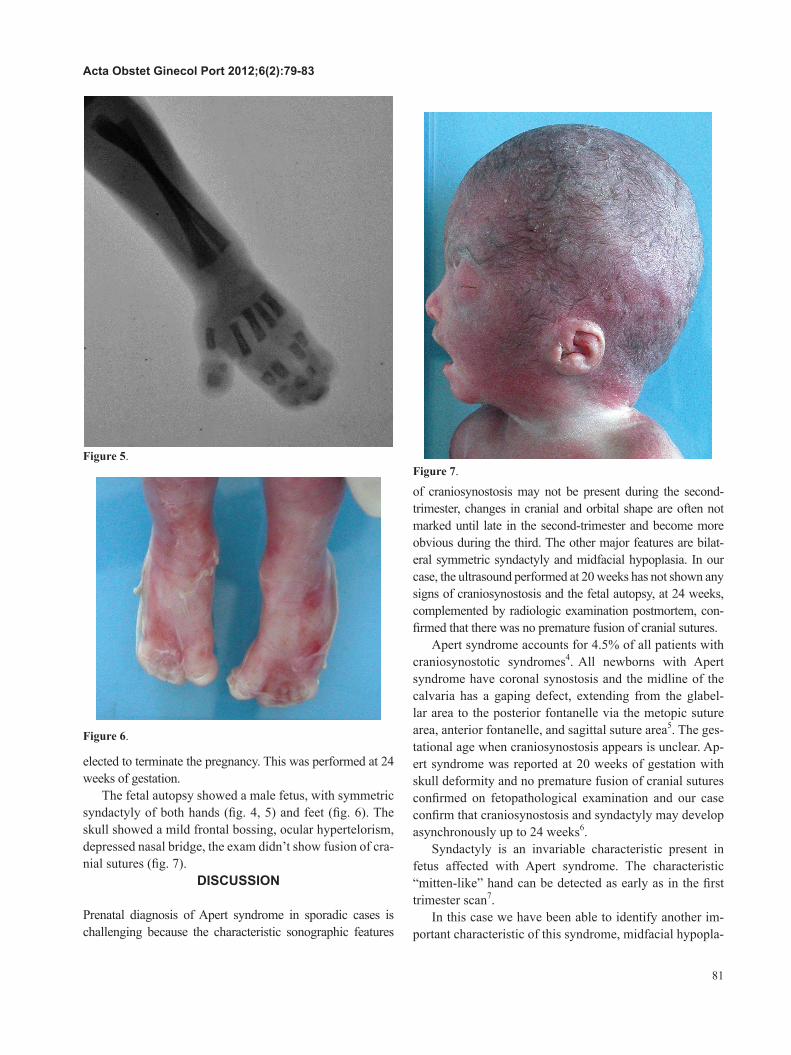
The fetal autopsy showed a male fetus, with symmetric syndactyly of both hands (ig. 4, 5) and feet (ig. 6). The skull showed a mild frontal bossing, ocular hypertelorism, depressed nasal bridge, the exam didn’t show fusion of cra-nial sutures (ig. 7).
diScuSSion
Prenatal diagnosis of Apert syndrome in sporadic cases is challenging because the characteristic sonographic features
of craniosynostosis may not be present during the second-trimester, changes in cranial and orbital shape are often not marked until late in the second-trimester and become more obvious during the third. The other major features are bilat-eral symmetric syndactyly and midfacial hypoplasia. In our case, the ultrasound performed at 20 weeks has not shown any signs of craniosynostosis and the fetal autopsy, at 24 weeks, complemented by radiologic examination postmortem, con-irmed that there was no premature fusion of cranial sutures.
Apert syndrome accounts for 4.5% of all patients with craniosynostotic syndromes4. All newborns with Apert syndrome have coronal synostosis and the midline of the calvaria has a gaping defect, extending from the glabel-lar area to the posterior fontanelle via the metopic suture area, anterior fontanelle, and sagittal suture area5. The ges-tational age when craniosynostosis appears is unclear. Ap-ert syndrome was reported at 20 weeks of gestation with skull deformity and no premature fusion of cranial sutures conirmed on fetopathological examination and our case conirm that craniosynostosis and syndactyly may develop asynchronously up to 24 weeks6.
Syndactyly is an invariable characteristic present in fetus affected with Apert syndrome. The characteristic “mitten-like” hand can be detected as early as in the irst trimester scan7.
In this case we have been able to identify another im-portant characteristic of this syndrome, midfacial hypopla-
Figure 5.
Figure 6.
Figure 7.
acta obstet ginecol port 2012;6(2):79-83

82
sia, inferring it by the presence of hypertelorism.This is the third case of Apert syndrome reported asso-
ciated with increased NT, the accumulation of nuchal luid may be caused by the altered composition of the extracel-lular matrix, as a consequence of altered conformation of the FGFR2 protein8, 9.
Although there were no cerebral anomalies detected, central nervous system anomalies have been frequently re-ported: non-progressive ventriculomegaly, hydrocephalus, partial absence of septum pellucidum and partial or com-plete agenesis of the corpus callosum10.
Several other syndromes that include craniosynostosis can lead to a similar appearance of the face and head, but do not include the severe hand and foot problems of Ap-ert syndrome. These similar syndromes include: Carpenter syndrome (kleeblattschadel, cloverleaf skull deformity), Crouzon disease (craniofacial dysostosis), Pfeiffer syn-drome and Saethre-Chotzen syndrome11.
When Apert syndrome was suspected the diagnosis was conirmed by DNA analysis for the S252W and P253R mutations in the FGFR2 gene, which together account for 98% of this type of craniosynostosis12. The other muta-tions involved the insertion of Alu-element mutations in or near exon 9 of FGFR2. DNA extracted from the amnio-cytes showed a S252W mutation. This is the most common mutation, occurring in 67% of patients and is associated with more severe craniofacial anomalies13. The majority of cases are due to de novo mutations which are increased exponentially with paternal age. In this case, the father was 35 years-old and in a population-based study almost half of fathers were older than 35 years when the child was born14.
It’s important to give accurate information about the prognosis of the affected individuals. An important question that should be considered in prenatal counseling is the possi-bility of mental retardation. Although patients of normal in-telligence have been reported approximately one half of the affected individuals are mentally retarded15, 16. The majority will require multiple surgeries, like craniofacial disjunction or shunting to reduce intracranial pressure, and to correct syndactyly9, 11. Despite multiple reconstructive procedures will be necessary, they will play an important role in enhanc-ing the psychosocial condition of the patient17.
Considering the vast majority of the cases are sporadic (>98%), the parents should be informed that the risk of re-currence is minimal but autosomal dominant inheritance and germinal mosaicism have been reported and should also be taken into consideration in genetic counseling13.
Apert syndrome is being diagnosed at early gestational ages using ultrasound and DNA analysis. This case dem-
onstrates the feasibility of the prenatal diagnosis of Apert syndrome and demonstrates that syndactyly and abnormal-ities of skull shape should lead to the suspicion of Apert syndrome even in the absence of craniosynostosis.
reFerenceS
1. Moloney DM, Slaney SF, Oldridge M, Wall SA, Sahlin P, Stenman G, Wilkie AO. Exclusive paternal origin of new mutations in Apert syndrome. Nat Genet. 1996 May;13(1):48-53.
2. Cohen MM Jr, Kreiborg S, Lammer EJ, Cordero JF, et al. Birth prevalence study of the Apert syndrome. Am J Med Genet. Mar 1 1992;42(5):655-9.
3. Wilkie AO, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, hockley AD, hayward RD, David DJ, Pulleyn LJ, Rutland P, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995 Feb;9(2):165-72.
4. Patel B, Suchet I. Postmortem computed tomography in a case of apert syndrome: correlation with conventional autopsy and prenatal ultrasound. ultrasound Q. 2010 Dec;26(4):249-53.
5. Kreiborg S, Marsh JL, Cohen MM Jr, Liversage M, Pedersen h, Skovby F, Borgesen SE, Vannier MW. Comparative three-dimensional analysis of CT scans of the calvaria and cranial base in Apert and Crouzon syndromes. J Craniomaxillofac Surg. 1993;21:181-188
6. Lyu KJ, Ko TM. Prenatal diagnosis of Apert syndrome with widely separated cranial sutures. Prenat Diagn. 2000 Mar;20(3):254-6.
7. Filkins K, Russo JF, Boehmer S, Camous M, Przylepa KA, Jiang W, Jabs EW. Prenatal ultrasonographic and molecular diagnosis of Apert syndrome. Prenat Diagn. 1997 Nov;17(11):1081-4.
8. Aleem S, howarth ES. Apert syndrome associated with increased fetal nuchal translucency. Prenat Diagn. 2005 Nov;25(11):1066-7.
9. David AL, Turnbull C, Scott R, Freeman J, Bilardo CM, van Maarle M, Chitty LS. Diagnosis of Apert syndrome in the second-trimester using 2D and 3D ultrasound. Prenat Diagn. 2007 Jul;27(7):629-32.
10. Quintero-Rivera F, Robson CD, Reiss RE, Levine D, Benson C, Mulliken JB, Kimonis VE. Apert syndrome: what prenatal radiographic indings should prompt its consideration? Prenat Diagn. 2006 Oct;26(10):966-72.
11. Kinsman SL, Johnston MV. Congenital Anomalies of the Central Nervous System. In: Nelson Textbook of Pediatrics. (18th Edition). Kliegman RM, Behrman RE, Jenson hB, Stanton BF (eds). Saunders Elsevier; 2007: chap 592.
12. Park WJ, Theda C, Maestri NE, Meyers GA, Fryburg JS, Dufresne C, Cohen MM Jr, Jabs EW. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J hum Genet. 1995 Aug;57(2):321-8.
13. Athanasiadis AP, zafrakas M, Polychronou P, Florentin-Arar L, Papasozomenou P, Norbury G, Bontis JN. Apert syndrome: the current role of prenatal ultrasound and genetic analysis in diagnosis and counselling. Fetal Diagn Ther. 2008;24(4):495-8.
14. Tolarova MM, harris JA, Ordway DE, Vargervik K. Birth prevalence, mutation rate, sex ratio, parents’ age, and ethnicity in Apert syndrome. Am J Med Genet. 1997 Nov 12;72(4):394-8.
15. Skidmore DL, Pai AP, Toi A, Steele L, Chitayat D. Prenatal diagnosis of Apert syndrome: report of two cases. Prenat Diagn. 2003 Dec 15;23(12):1009-13.
16. Esser T, Rogalla P, Bamberg C, Kalache KD. Application of the three-dimensional maximum mode in prenatal diagnosis of Apert syndrome. Am J Obstet Gynecol. 2005 Nov;193(5):1743-5.
17. Allam KA, Wan DC, Khwanngern K, Kawamoto hK, Tanna N, Perry A, Bradley JP. Treatment of Apert Syndrome: A Long-term Follow-up Study. Plast Reconstr Surg. 2010 Dec 23
azevedo H, pereira e, anderson J, rosmaninho a, Barbêdo c