patologias embriologicas de genitales
TRANSCRIPT

PATOLOGIA DE APARATO
GENITAL

MALFORMACIONES DE
ÚTERO Y VAGINA

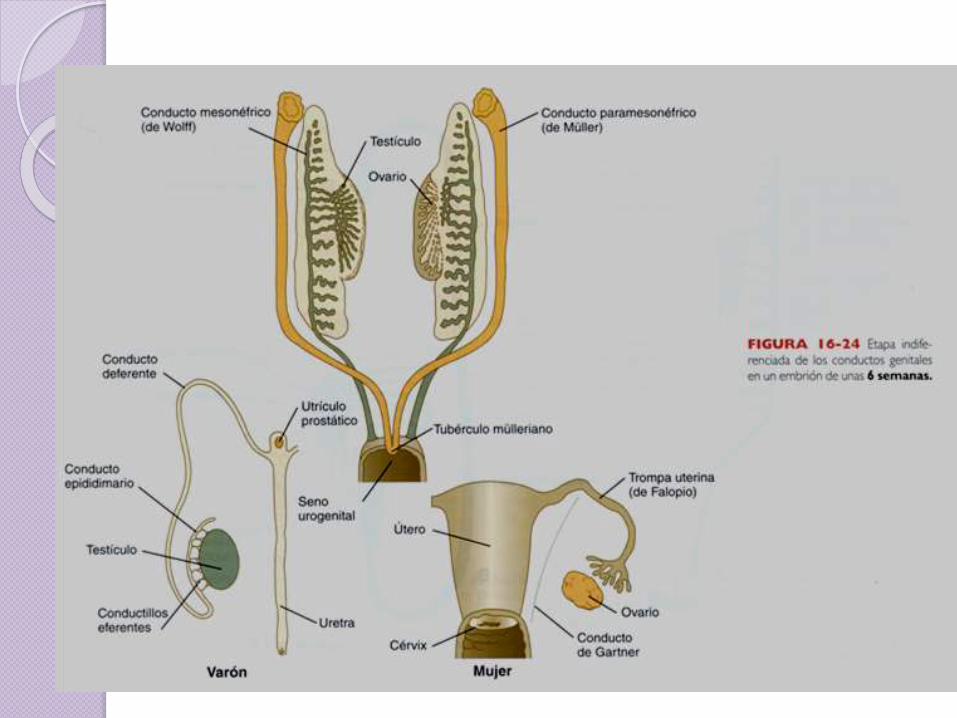
Conductos de Müller
Aparición en cresta urogenital
y progresión hacia cloaca:
semana 6-9
Fusión (conducto
uretrovaginal): Semana 10-13
Reabsorción de tabique:
semana 14-16

C. Müller = útero + trompas + 1/3
sup vagina.
Interrupción semana 6-9 = aplasia
Interrupción semana 9-13=
defectos de fusión (útero bicorne)
Interrupción semana 14-18 =
defectos de reabsorción (útero
septo)
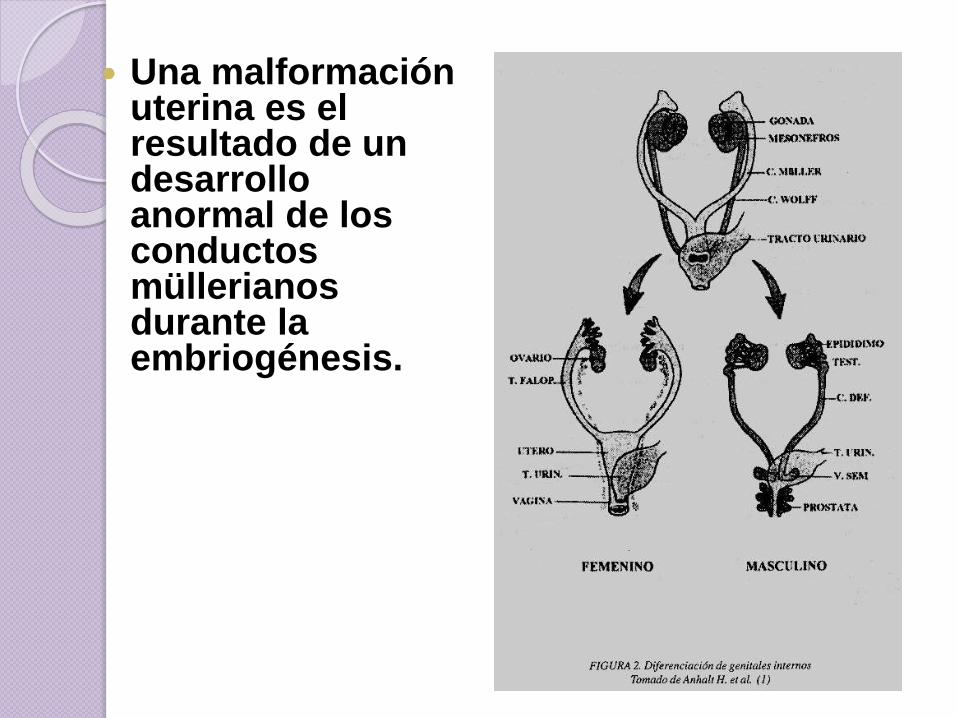
Una malformación uterina es el resultado de un desarrollo anormal de los conductos müllerianosdurante la embriogénesis.
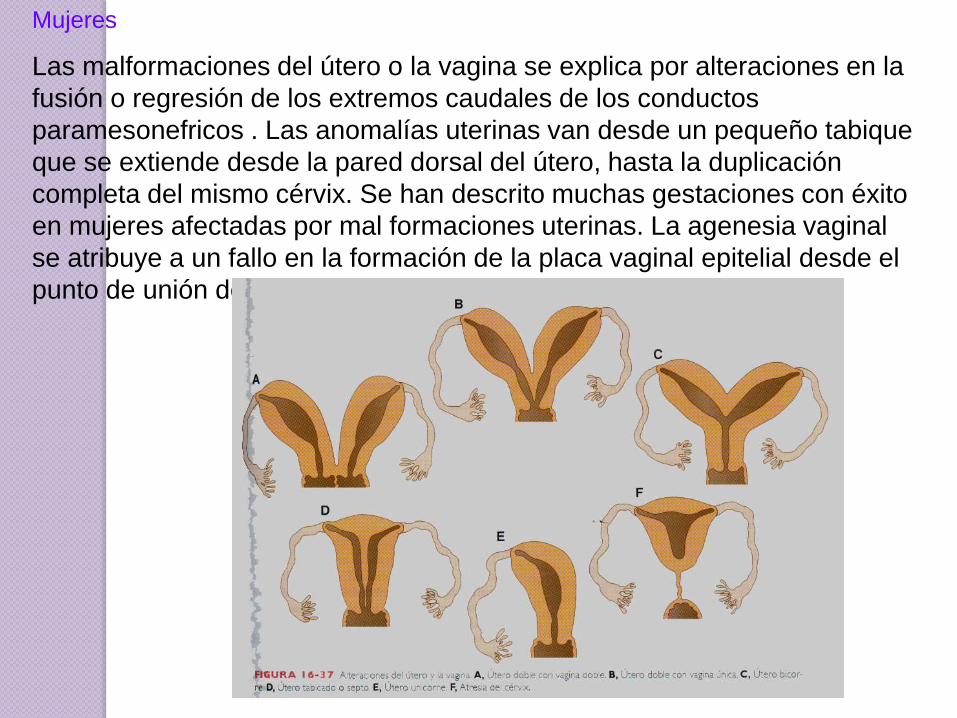
Mujeres
Las malformaciones del útero o la vagina se explica por alteraciones en la
fusión o regresión de los extremos caudales de los conductos
paramesonefricos . Las anomalías uterinas van desde un pequeño tabique
que se extiende desde la pared dorsal del útero, hasta la duplicación
completa del mismo cérvix. Se han descrito muchas gestaciones con éxito
en mujeres afectadas por mal formaciones uterinas. La agenesia vaginal
se atribuye a un fallo en la formación de la placa vaginal epitelial desde el
punto de unión del tubérculo mülleriano con el seno urogenital.
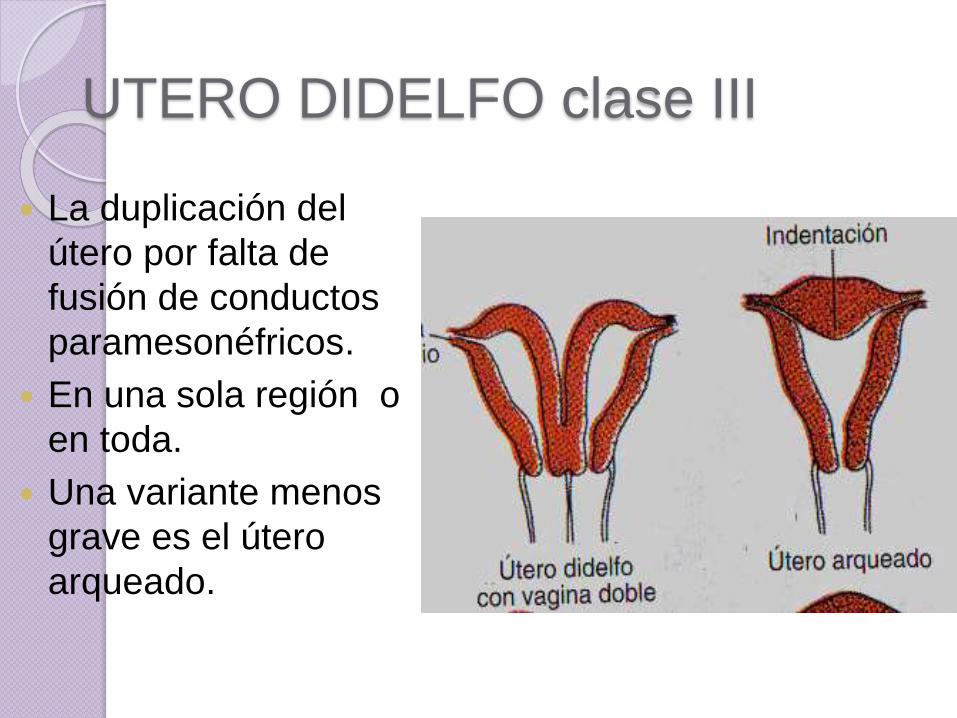
UTERO DIDELFO clase III
La duplicación del
útero por falta de
fusión de conductos
paramesonéfricos.
En una sola región o
en toda.
Una variante menos
grave es el útero
arqueado.
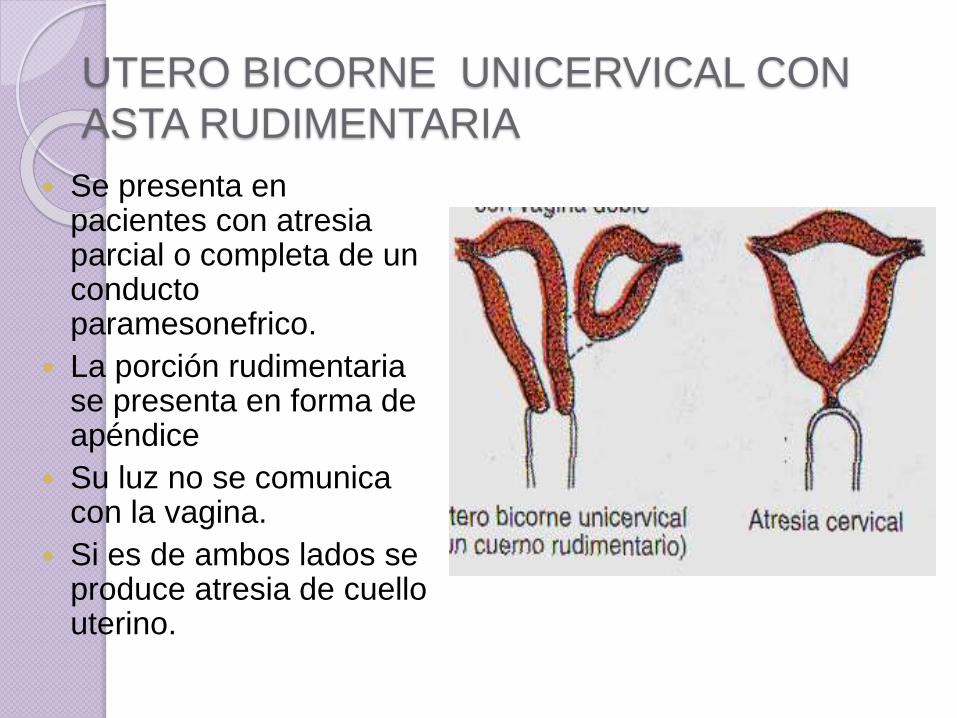
UTERO BICORNE UNICERVICAL CON
ASTA RUDIMENTARIA
Se presenta en pacientes con atresia parcial o completa de un conducto paramesonefrico.
La porción rudimentaria se presenta en forma de apéndice
Su luz no se comunica con la vagina.
Si es de ambos lados se produce atresia de cuello uterino.

La clasificación de la Asociación Americana de Medicina Reproductiva distingue los siguientes tipos:
Clase I: Agenesia Mülleriana(ausencia de útero). El útero no está presente, vagina rudimentaria o ausente. Esta condición también es llamada síndrome Mayer-Rokitansky-Kuster-Hauser. La paciente con síndrome MRKH tendrá amenorrea primaria.
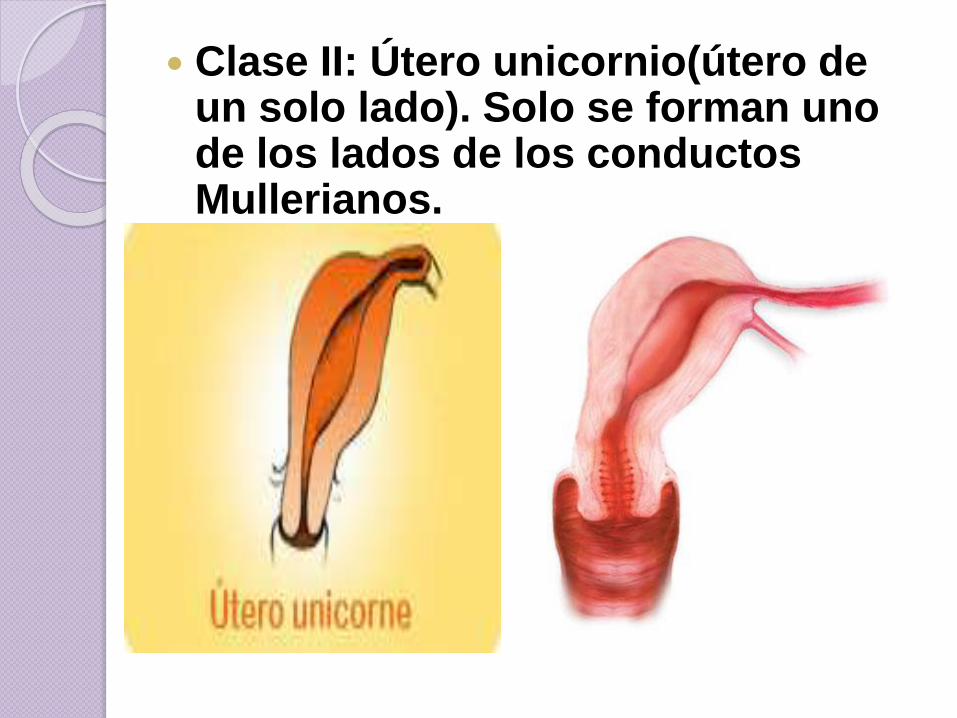
Clase II: Útero unicornio(útero de un solo lado). Solo se forman uno de los lados de los conductos Mullerianos.
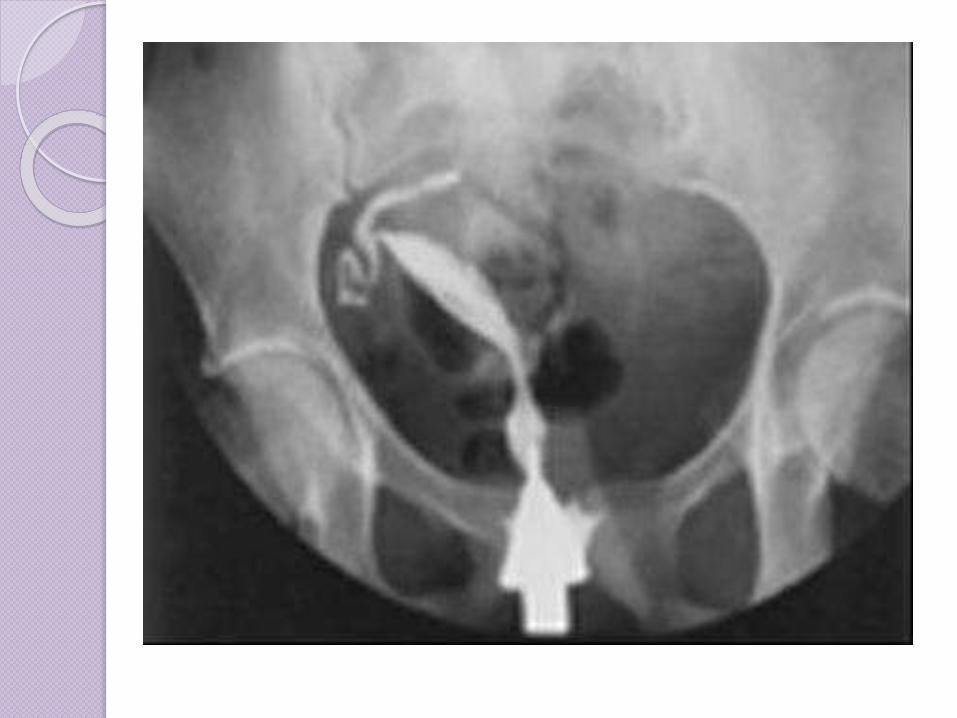

Clase III: Útero didelfo(doble
útero). Ambos conductos
Mullerianos se desarrollan pero
no se llegan a fusionar, por
ende la paciente tiene un
“doble útero”. En esta
condición pueden darse un
doble cérvix, una partición
vaginal o embarazos
espontáneos en ambos úteros.
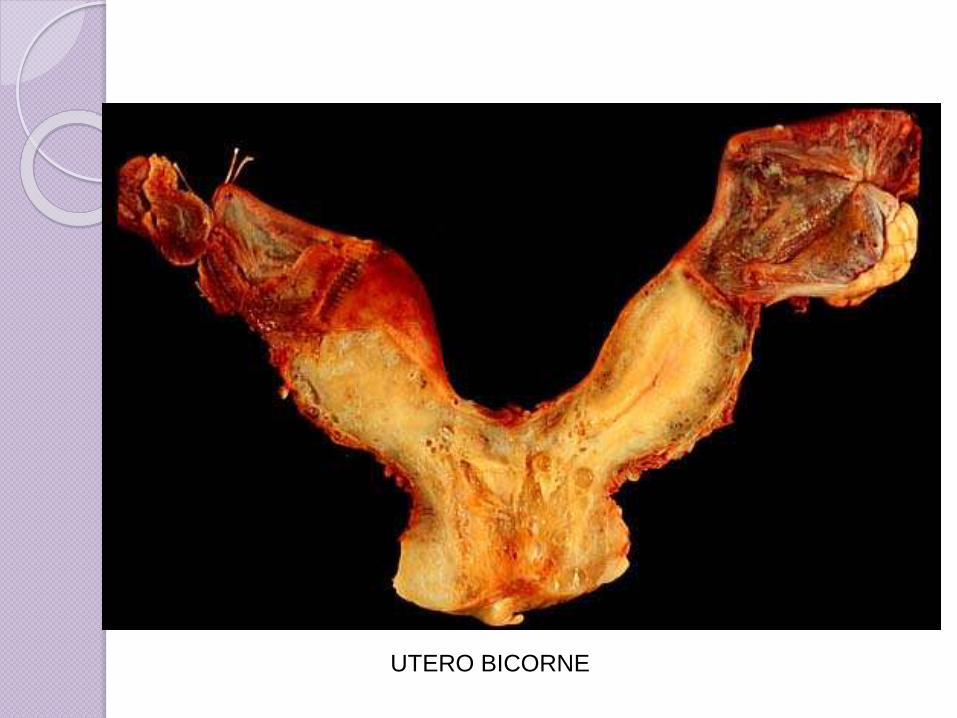
UTERO BICORNE

Clase IV: Útero Bicornio(útero
con dos cuernos). Solo la parte
superior del sistema Mulleriano
no se fusiona dando como
resultado que la parte caudal
del útero sea normal. La parte
craneal del útero está
bifurcada provocando así que
el útero tenga “forma de
corazón”.

Clase IV. 10% de
malformaciones
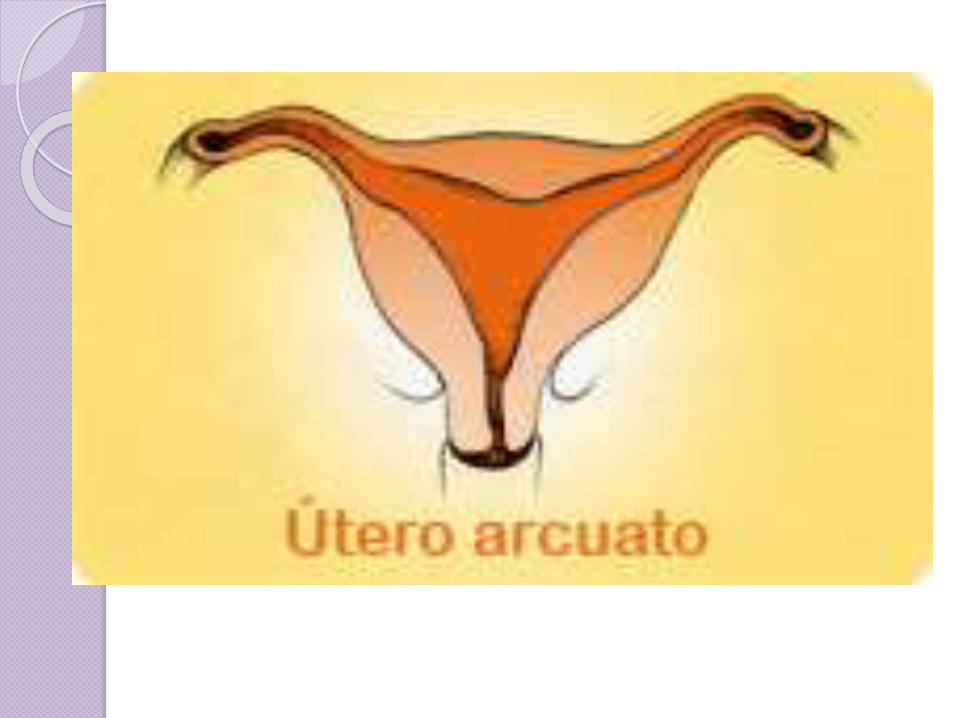
Útero arcuato

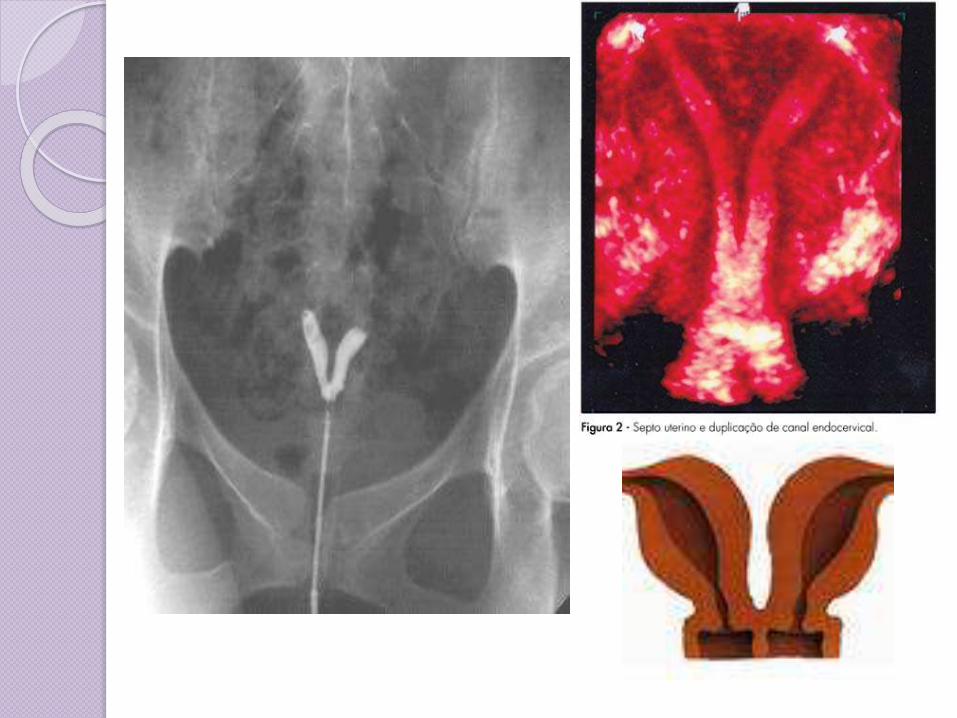
Clase V: Útero particionado (Septum uterino o partición). Los dos conductos Mullerianosestán fusionados pero la separación entre ellos todavía está presente, separando el sistema en dos partes. Un septum uterino es la malformación uterina más común y la causa de muchos abortos espontáneos.

Clase VI: Útero dietilstibestrol(DES).
◦La cavidad uterina tiene “forma de T” como resultado de la exposición fetal al dietilstilbestrol.
Una variación adicional es el útero acuarto donde hay un hoyuelo cóncavo en el fondo uterino dentro de la cavidad.


Un útero rudimentario son los restos uterinos no conectados con el cérvix y la vagina y pueden ser encontrados al otro lado de un útero unicornio.
Pacientes con anormalidades uterinas pueden tener anormalidades renales asociadas incluyendo agénesis unilateral renal

“Doble Vagina”
La vagina se deriva de los
conductos Mullerianos; la no
fusión de estos dos conductos
puede ocasionar la formación
de una duplicación vaginal y
poca absorción de la pared
entre los dos conductos puede
dejar un septum residual, lo
que lleva a una “doble vagina”.
Si es necesario, la partición
puede ser corregida


Diagnostico
Aparte del examen físico, se necesita tomar imágenes para determinar las características de la malformación: Ultrasonografía ginecológica, resonancia magnética pélvica o histerosalpingografía. Un histerosalpingograma no es considerado de utilidad debido a su inhabilidad para evaluar el contorno exterior del útero y distinguir entre un bicornio y un útero particionado. Adicionalmente una laparoscopia y/o histeroscopia pueden ser recetados.

La mayoría asintomática
Frecuencia 6.7 % de la población
Infértiles 3-3.5% , 16% en útero arcuato
Estériles 2.3%
Multifactorial
Alteraciones genéticas trisomia 18-13, monosomia X, translocacion 9-17
Causas yatrogenas: talidomida
Alcoholismo

Malformaciones Renales
Frecuencia: 30%
Frecuencia de malformaciones
uterinas en agenesia renales:
55-70 %
Si los teratogénos actúan entre
la semana 6-9
Se asocia con malformaciones
vaginales, anales y óseas.

ALTERACIONES GENITALES MASCULINOS

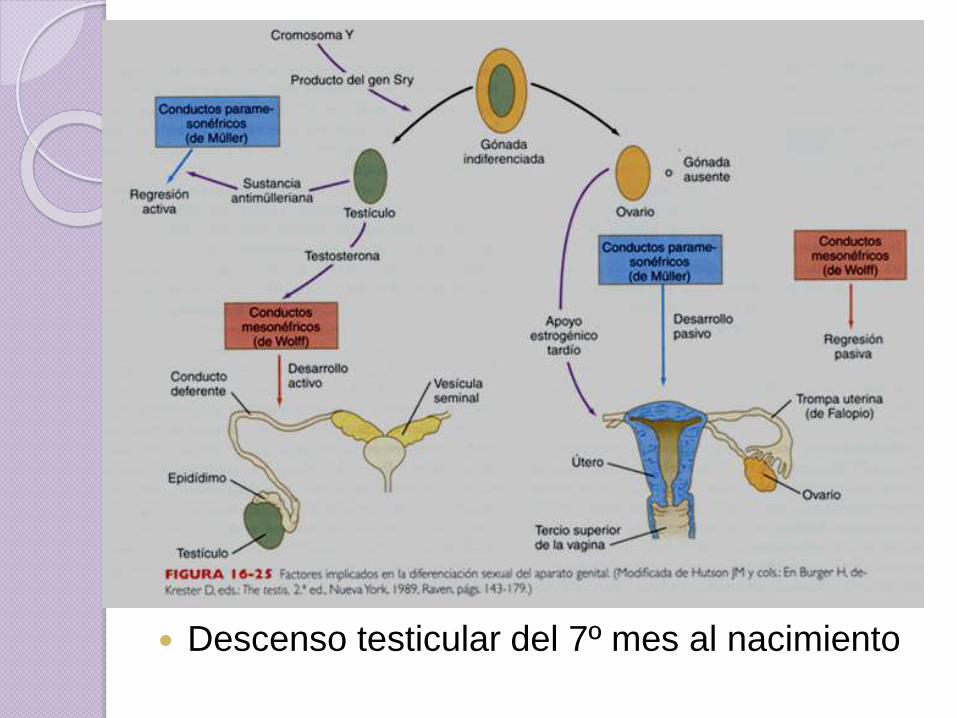
Descenso testicular del 7º mes al nacimiento

Criptorquidia El 3% de los niños nacidos de término y el 30% de los
prematuros nacen sin que uno o sus dos testículos hayan descendido al escroto, lo que se conoce como criptorquidia unilateral o bilateral.
El defecto evoluciona a la normalidad en la mayoría de los casos:
la mitad, en el primer mes postnatal,
25% en el transcurso del primer año.
Durante este período, el médico debe mantener una actitud vigilante y localizar la situación de los testículos, lo que le permitirá un diagnóstico más preciso, descartando las posibilidades de testículo retráctil, testículo ectópico (fuera del conducto inguinal) y de agenesias testicular o anorquia.
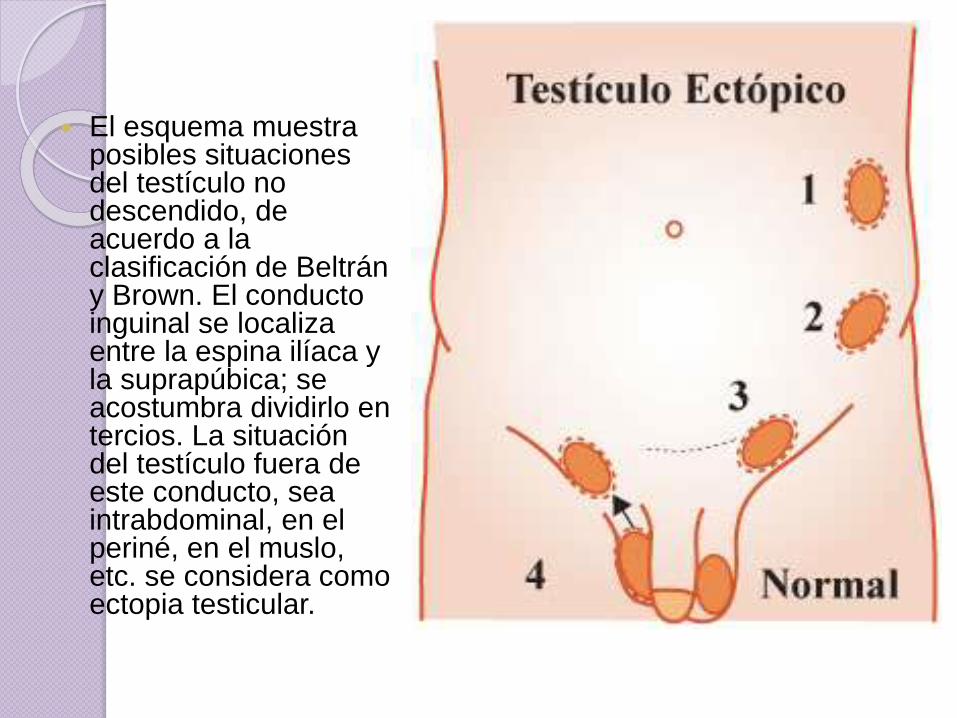
El esquema muestra posibles situaciones del testículo no descendido, de acuerdo a la clasificación de Beltrán y Brown. El conducto inguinal se localiza entre la espina ilíaca y la suprapúbica; se acostumbra dividirlo en tercios. La situación del testículo fuera de este conducto, sea intrabdominal, en el periné, en el muslo, etc. se considera como ectopia testicular.
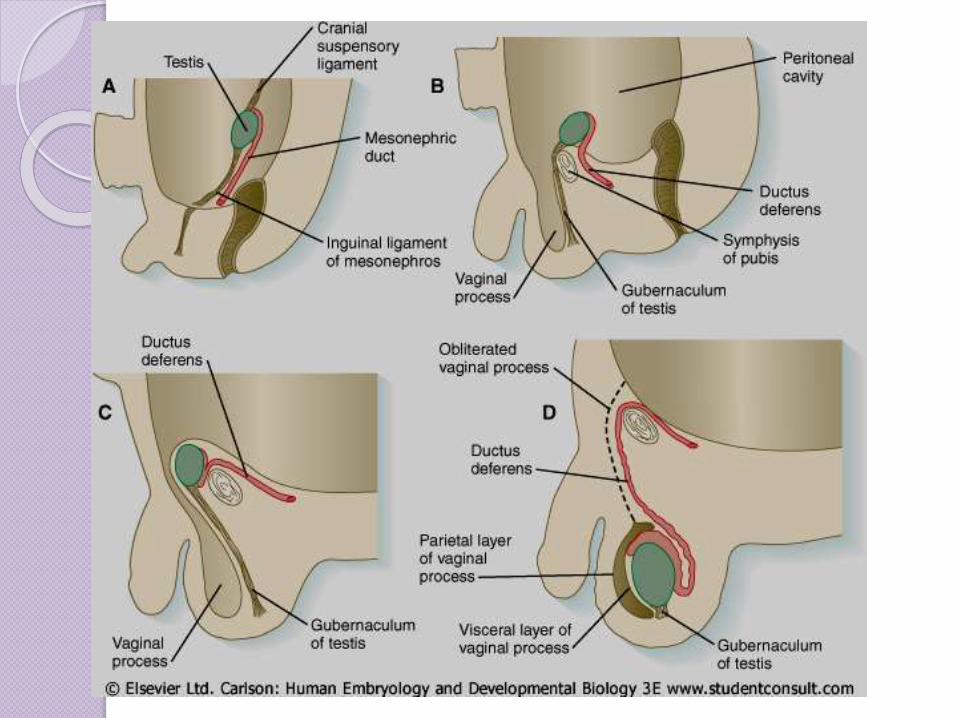

El tratamiento de la criptorquidia es
quirúrgico y debe hacerse entre los 4
y 5 años de edad, ya que la
temperatura intrabdominal daña el
tejido reproductor y aumenta las
posibilidades de malignización
testicular.

El 4% de los niños con testículos no descendido presenta una historia familiar en la que el caso se repite; es concordante en 7 de 10 parejas de gemelos idénticos; se asocia a síndromes XXXY, mosaicos XO/XY, XO/XY/XX, trisomías 13-15, algunos casos XYY y con el 50% de los varones con síndrome de Down, no siendo raro en otros síndromes donde hay marcado retardo del desarrollo intrauterino.

Anorquia congénita
Se estima que ocurre en 1:20.000 nacidos vivos.
Es la destrucción de la gónada masculina que se
produce después de la semana 12-14 cuando se a
completado la diferenciación de los genitales externos.
El bebé exhibirá genitales masculinos externos e
internos normales (pene y escroto), pero se presentará
ausencia de testículos. Esta afección se conoce como
anorquia congénita.
Debe efectuarse una prueba de estimulación con
gonadotropina coriónica humana (prueba de hCG) para
distinguir entre anorquía (-) y criptorquidia.

anorquia congénita
Entre los signos están: Escroto vacío Ausencia de características sexuales
secundarias Entre los exámenes están: Niveles de gonadotropina (negativos) Niveles elevados de hormona
folículoestimulante y hormona luteinizante
Ecografía o IRM que muestra ausencia de tejido gonadal
Densidad ósea baja.

anorquia congénita

MICROPENE
No hay estimulo suficiente androgénico.
Es por hipogonadismo primario.
O por disfunción hipotalamohipofisiaria.
Tiene una longitud de 2.5 por debajo de lo normal.
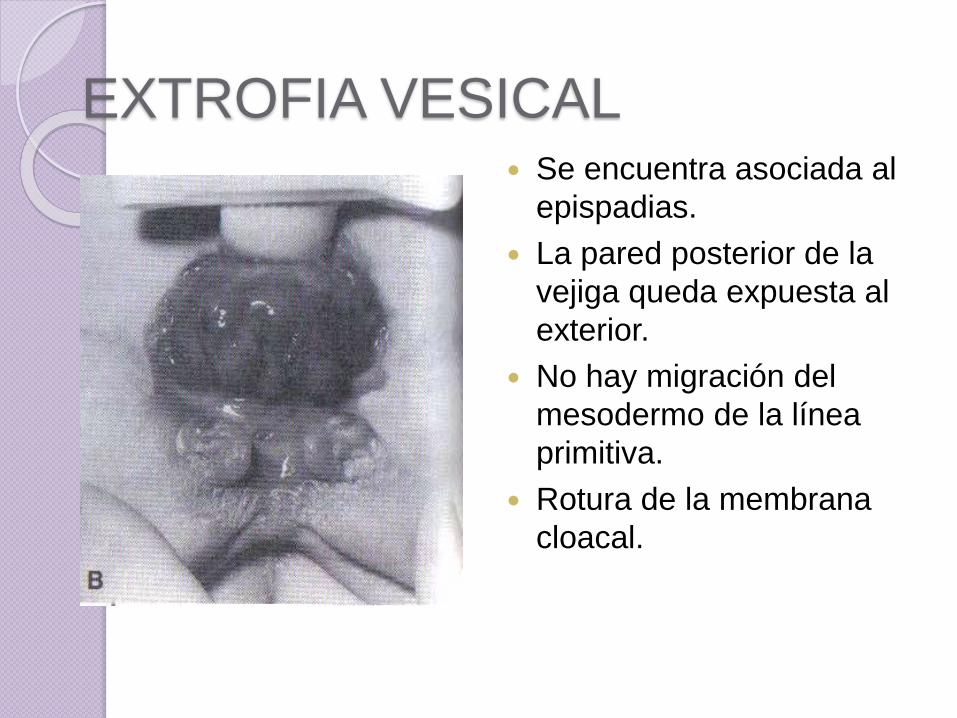
EXTROFIA VESICAL Se encuentra asociada al
epispadias.
La pared posterior de la
vejiga queda expuesta al
exterior.
No hay migración del
mesodermo de la línea
primitiva.
Rotura de la membrana
cloacal.

EPISPADIAS
La uretra esta en el dorso del pene.
El tubérculo genital se formo en la región del tabique urorrectal.
Se acompaña de extrofia vesical.

EPISPADIAS
Causas, incidencia y factores de riesgo
Las causas del epispadias se desconocen en este momento, pero se cree que está relacionado con el desarrollo inadecuado del hueso púbico.
En los niños con epispadias, la abertura de la uretra generalmente se encuentra en el extremo o al lado del pene, en lugar de estar en la punta. Sin embargo, es posible que la uretra esté abierta a lo largo de todo el pene.

Epispadias: Incidencia
El epispadias se presenta en 1 de cada
177.000 niños varones y en una de
484.000 niñas recién nacidos. Esta
afección generalmente se diagnostica en
el momento del nacimiento o poco
después.
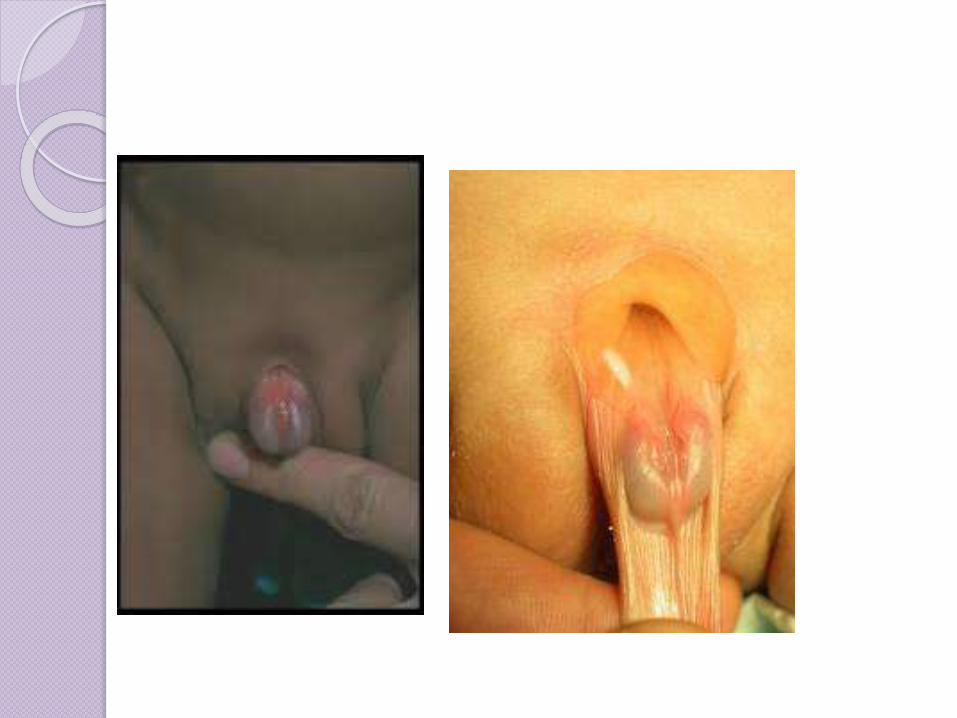

Epispadias
Síntomas
Abertura anormal desde la articulación entre el hueso púbico hasta el área por encima de la punta del pene
Reflujo de la orina hacia el riñón (nefropatía por reflujo)
Pene corto y ensanchado con una curvatura anormal
Infecciones urinarias
Ensanchamiento del hueso púbico
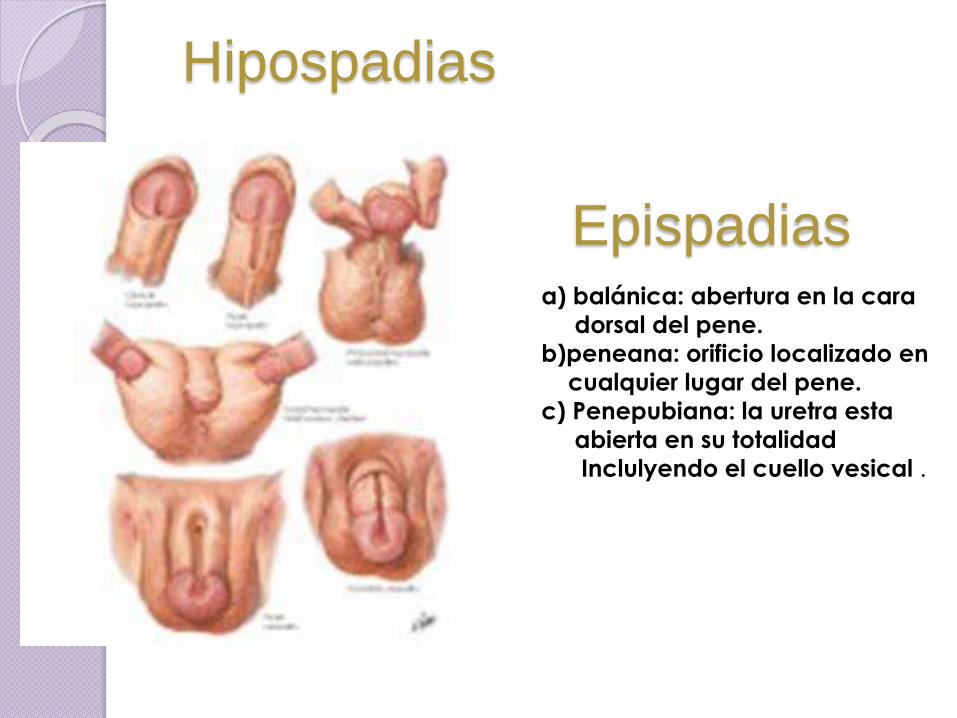
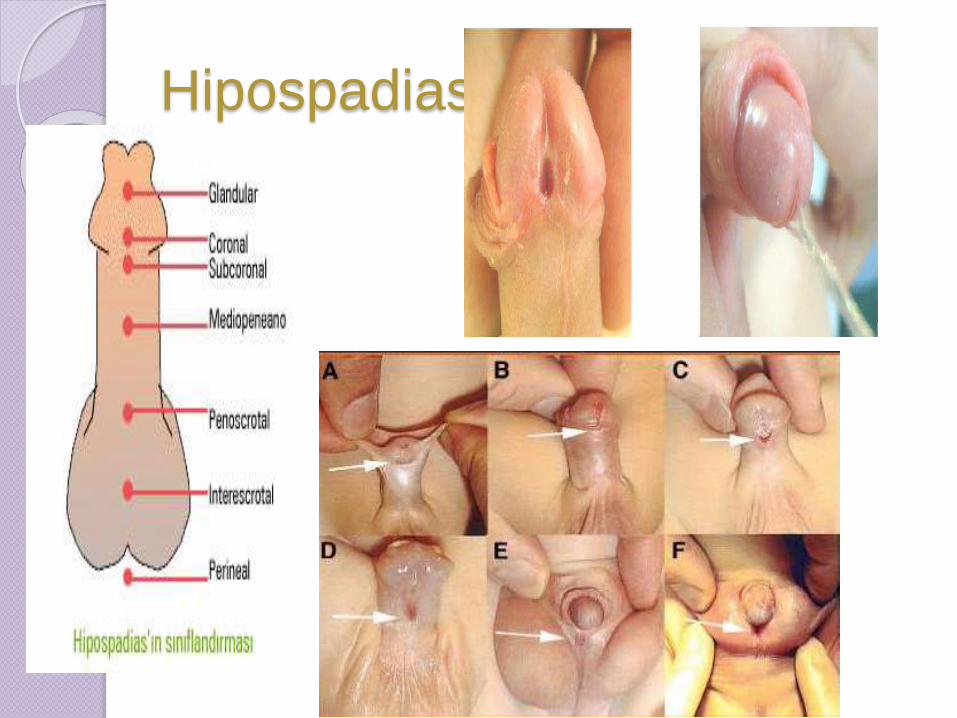
Hipospadias
a) balánica: abertura en la cara
dorsal del pene.
b)peneana: orificio localizado en
cualquier lugar del pene.
c) Penepubiana: la uretra esta
abierta en su totalidad
Inclulyendo el cuello vesical .
Epispadias

Exámenes
Examen de sangre para verificar los niveles de electrolitos
Pielografía intravenosa (PIV), una radiografía especial de los riñones, la vejiga y los uréteres
Radiografía de la pelvis
Ecografía del sistema urogenital
Tratamiento
La reparación quirúrgica del epispadias se recomienda en pacientes con más que un caso leve. El escape de orina (incontinencia) no es común y puede requerir una segunda operación.

Pronóstico La cirugía generalmente lleva a la
capacidad para controlar el flujo de orina y a un buen resultado estético.
Complicaciones Se puede presentar incontinencia
urinaria persistente en algunas personas con esta afección, aun después de la cirugia.
Asimismo, puede ocurrir daño en las vías urinarias (uréter y riñón) al igual que esterilidad.

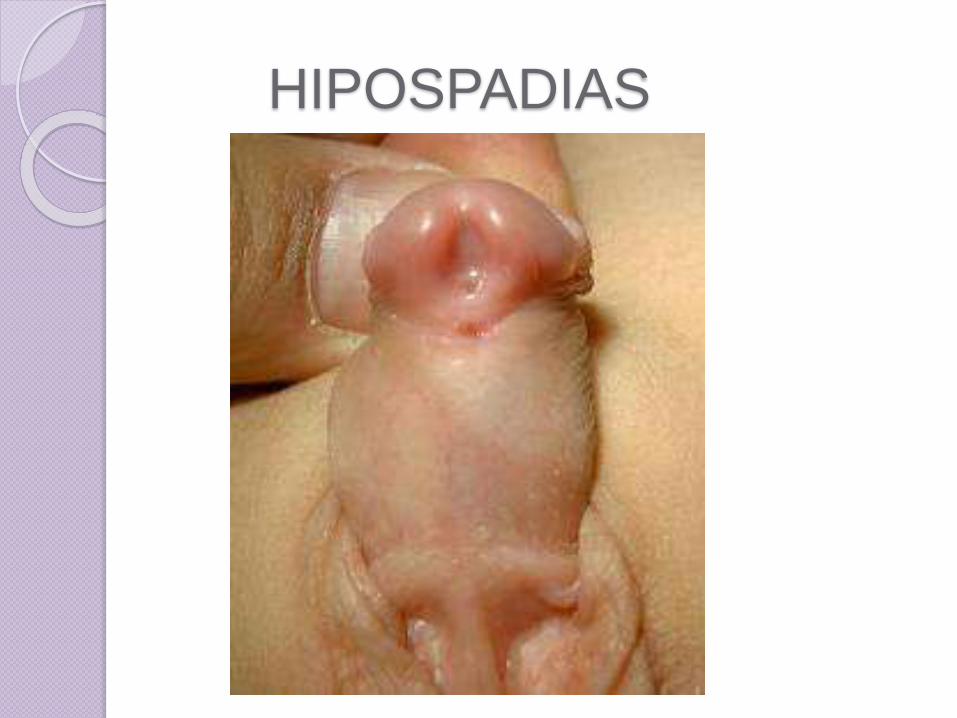
Hipospadias
• El hipospadias afecta 4 de cada 1,000varones recién nacidos
• Es una anomalía congénita en la que la seobserva una abertura del pene, el meatourinario se localiza en algún lugar en la parteinferior del glande o cuerpo del pene, o másatrás, como en la unión del escroto y pene.
• Esta malformación es debida a una fusiónincompleta de los pliegues uretrales.
• Además, el prepucio no se desarrollatotalmente, sino que forma una capuchasobre el del glande, con adherencias.
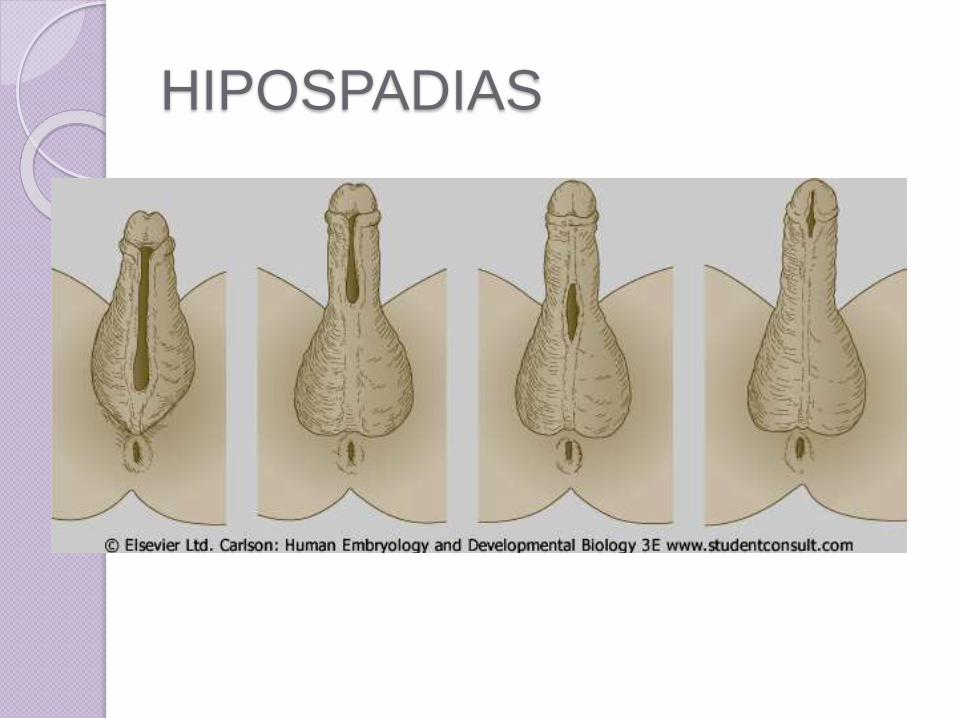
HIPOSPADIAS
HIPOSPADIAS
Hipospadias

OTRAS ALTERACIONES DEL SISTEMA DE CONDUCTOS GENITALES
Varones
Las anomalías del sistema de conductos mesonefricos son
relativamente infrecuentes, pero se pueden producir duplicaciones o
divertículos del conducto deferente o la uretra. Existe una interesante
correlación entre la ausencia de conducto deferente o su persistencia
rudimentaria en los varones y la fibrosis quística. Esta asociación
puede ser consecuencia de un defecto en un gen situado al lado del
que produce dicha fibrosis quística.
El síndrome de persistencia del conducto mulleriano se caracteriza por
la formación de trompas de Falopio y de un útero y se ha descrito en
algunos varones 46 XY. No Existe una causa única para esta alteración
y se han demostrado mutaciones de los genes que codifican la
sustancia antimulleriana y su receptor.